






Índice
- Por que a ciência dos cannabinoid não pode ser reduzida a CB1 e CB2
- O sistema endocannabinoid versus o panorama mais amplo de alvos dos cannabinoid
- Canais TRP: os sensores de calor, dor e irritação que os cannabinoid continuam atingindo
- PPARs: cannabinoid como sinais lipídicos intracelulares, e não apenas ligantes de receptores de membrana
- GPR55, GPR18 e GPR119 e o problema dos GPCR órfãos
- Sinalização de serotonina: onde os cannabinoid se cruzam com os sistemas 5-HT
- Além da lista solicitada: canais de sódio e outros alvos não canônicos que já estão mudando a conversa sobre dor
- Como cannabinoid específicos diferem quando você deixa de perguntar apenas sobre CB1 e CB2
- Os métodos importam: por que o desenho do ensaio molda o que pensamos que os cannabinoid fazem
- Níveis de evidência: da placa de células à clínica
- Segurança, regulação e por que a farmacologia fora do alvo importa em saúde pública
- Descoberta de fármacos: projetando cannabinoid e moléculas inspiradas em cannabinoid para alvos não CB1/CB2
- Equívocos comuns e controvérsias não resolvidas
- Interpretação prática para leitores, clínicos e pesquisadores
Por que a ciência dos cannabinoid não pode ser reduzida a CB1 e CB2
A versão resumida da farmacologia dos cannabinoid diz o seguinte: THC atua em CB1, os efeitos imunológicos passam por CB2, e todo o resto é um rodapé. Essa formulação é fácil de ensinar e fácil de repetir. Também está errada com frequência suficiente para bloquear uma compreensão séria de dor, inflamação, ansiedade, prurido, náusea, metabolismo e neuroproteção.
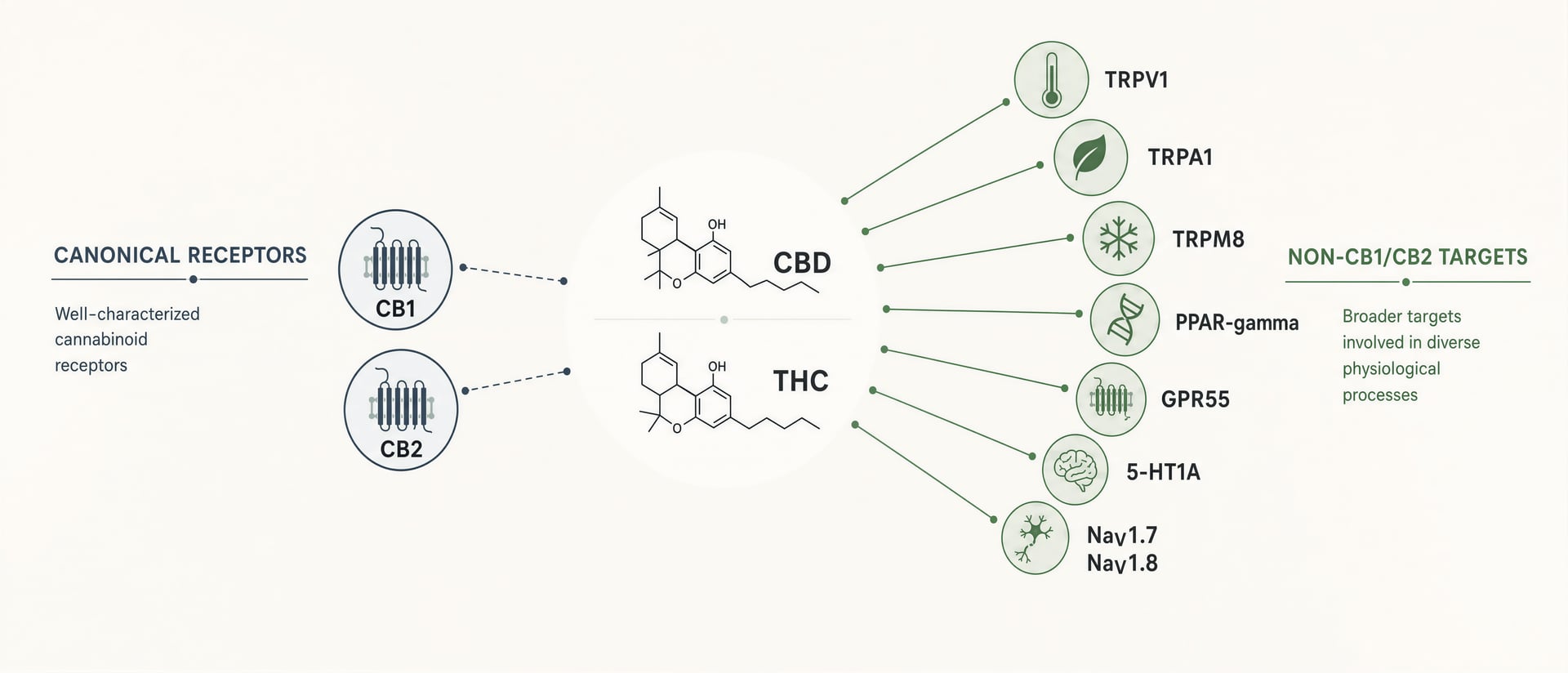
CB1 e CB2 importam. CB1 é abundante no cérebro e explica grande parte da intoxicação causada por THC, da alteração da memória, dos efeitos sobre o apetite e de parte de sua analgesia. CB2 é central para muitas discussões sobre imunidade e inflamação. Mas os cannabinoid não são ligantes arrumados, feitos para um receptor cada um. São moléculas lipofílicas e flexíveis, que interagem com um campo farmacológico mais amplo: canais de potencial receptor transitório, como TRPV1 e TRPA1; receptores nucleares, como PPAR-gamma; GPCR órfãos ou ainda debatidos relacionados aos cannabinoid, como GPR55 e GPR18; receptores de serotonina, incluindo 5-HT1A; sinalização relacionada à adenosina; transporte e metabolismo de ácidos graxos; e, em trabalhos mais recentes, canais de sódio dependentes de voltagem, incluindo NaV1.7 e NaV1.8.[1]HHS and FDA support DEA action on dangerous 7-OH products. U.S. Department of Health and Human Services. HHS Press Room, 2025. https://www.hhs.gov/press-room/hhs-fda-support-dea-7-oh-scheduling.html
Esse campo mais amplo importa porque o mecanismo determina risco e benefício. Os reguladores já estão enfrentando esse problema em disputas adjacentes sobre políticas de drogas. Em 2025, o Department of Health and Human Services dos EUA afirmou que “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” ao apoiar a ação da DEA contra produtos com 7-OH intensificado. O composto específico não é um cannabinoid, mas a lição regulatória se transfere bem: quando químicos começam a modificar estruturas de produtos naturais e concentrar metabólitos, categorias simples baseadas na origem deixam de proteger o público. O perfil-alvo de uma molécula importa mais do que a forma como a escrita popular a trata como algo familiar.
O mito do receptor na escrita popular sobre cannabis
Os textos populares sobre cannabis costumam apresentar receptores como interruptores de liga/desliga: THC ativa CB1, CBD não “se liga fortemente”, logo CBD deve ser fraco ou misterioso. Esse relato funde várias ideias farmacológicas distintas em um verbo vago: ligar.
A agonismo ortostérico é o caso clássico. Um ligante ocupa o sítio ativo principal do receptor e estabiliza a sinalização. THC é um agonista parcial em CB1 e CB2. Esse é um tipo de ação, não o molde para toda a biologia dos cannabinoid. Um composto pode, em vez disso, agir alostericamente, alterando a forma como outro ligante funciona no receptor sem ocupar o mesmo sítio. Pode abrir, sensibilizar ou dessensibilizar um canal iônico. Pode entrar na célula e ativar um receptor nuclear que altera a transcrição gênica ao longo de horas, e não de milissegundos. Pode inibir um transportador, alterar propriedades de membrana ou retardar uma enzima que degrada um lipídio sinalizador endógeno.[2]EPIDIOLEX (cannabidiol) oral solution label. U.S. Food and Drug Administration. FDA drug label, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/210365s016lbl.pdf
CBD é a refutação mais clara do reducionismo de receptor. Seu uso clínico aprovado não se baseia em agonismo de CB1. O rótulo da FDA para solução oral de cannabidiol afirma que ela é indicada para convulsões associadas à síndrome de Lennox-Gastaut, à síndrome de Dravet e ao complexo de esclerose tuberosa em pacientes com 1 ano de idade ou mais. Qualquer que seja o mecanismo completo por trás desse efeito, ele não é adequadamente explicado pela antiga história de que ação cannabinoid relevante equivale a ativação forte de CB1 ou CB2. Entre os candidatos mecanísticos repetidamente citados na literatura estão TRPV1, sinalização relacionada a 5-HT1A, modulação da adenosina, efeitos intracelulares sobre cálcio e interações com enzimas ou transportadores. Nenhum pode ser tratado como a única resposta, mas, juntos, mostram por que o mito simplista do receptor falha.
A história também aponta na mesma direção. O trabalho de Raphael Mechoulam sobre endocannabinoid abriu um campo centrado em anandamida e 2-AG, mas até mesmo a anandamida não é apenas um ligante de CB1. Ela também ativa TRPV1, o receptor de calor e capsaicina cuja importância sensorial mais ampla foi reconhecida no Prêmio Nobel de 2021 concedido a David Julius e Ardem Patapoutian pelas descobertas de receptores para temperatura e toque. Uma vez que um endocannabinoid endógeno pode sinalizar tanto por um GPCR quanto por um canal TRP, o modelo “apenas CB1/CB2” deixa de ser um modelo. Vira uma caricatura.
Polifarmacologia: um ligante, muitos alvos
Um ponto de partida melhor é a polifarmacologia. Um ligante, muitos alvos, com diferentes afinidades, eficiências, tecidos e consequências. Em farmacologia, “sujo” às vezes é usado de modo pejorativo, mas, para os cannabinoid, muitas vezes é apenas descritivo.
Considere quantos tipos de ação cabem sob o mesmo termo-guarda-chuva. THC é um agonista parcial de CB1/CB2, mas trabalhos de 2025 destacados pela Hebrew University relataram que THC inibe nociceptores periféricos ao atingir os canais de sódio nociceptivos NaV1.7 e NaV1.8. Isso não é agonismo de receptor. É inibição de canal iônico em alvos já vistos como candidatos importantes para fármacos contra dor. Se essa linha de trabalho se mantiver em diferentes espécies e condições de dose, parte da analgesia do THC pode vir de um mecanismo que se parece mais com um freio local de excitabilidade do que com um efeito clássico de receptor cannabinoid.
CBD mostra um estilo diferente de promiscuidade. Em diferentes sistemas de ensaio, foi relatado que ele influencia TRPV1, TRPA1, TRPM8, 5-HT1A, PPAR-gamma, GPR55 e o tônus da adenosina, entre outros. O problema não é falta de mecanismos. O problema é separar quais mecanismos importam em concentrações atingidas clinicamente em humanos. O engajamento de alvo in vitro é barato. Traduzir isso é difícil. Um efeito em faixa micromolar, em uma linhagem celular que superexpressa o receptor, não explica automaticamente resultados em pacientes após dose oral, metabolismo de primeira passagem, ligação a proteínas e partição tecidual.
Outros phytocannabinoid complicam ainda mais o quadro. CBG já foi discutido como composto com interação alpha-2 adrenérgica, atividade em TRP e interação com 5-HT1A em alguns sistemas. CBC foi relacionado a TRPA1 e canais TRPV. THCV pode se comportar de modo diferente de delta-9-THC em CB1 dependendo da dose e do contexto, ao mesmo tempo em que mantém possibilidades fora de CB1. Cannabinoid ácidos, como CBDA e THCA, levantam perguntas adicionais porque descarboxilação, estabilidade e formação de metabólitos alteram a exposição ao alvo. O mesmo rótulo no frasco pode, portanto, esconder farmacologias muito diferentes quando a via de administração, o calor, o metabolismo e a formulação entram na história.[3]Library Docking for Cannabinoid-2 Receptor Ligands. American Chemical Society. Journal of Medicinal Chemistry, 2016. https://pubs.acs.org/doi/10.1021/acs.jmedchem.6c00835
Mesmo dentro da farmacologia de GPCR, o campo já passou de rótulos grosseiros. GPR55 ainda é às vezes chamado de candidato a “CB3”, mas isso continua contestado por um bom motivo; a sinalização, o conjunto de ligantes e o papel fisiológico não se encaixam de modo limpo nos receptores cannabinoid clássicos. GPR18 e GPR119 também aparecem na literatura adjacente aos cannabinoid, especialmente em inflamação, metabolismo e sinalização intestinal, mas as evidências são irregulares. Químicos medicinais conhecem isso. Um artigo de 2016 no Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, capturou uma abordagem estrutural que é quase o oposto da lore popular sobre receptores: desenho seletivo para o alvo, docking, otimização de scaffold e separação intencional dos efeitos desejados dos indesejados. O campo não está perguntando “isso atinge receptores cannabinoid?”; está perguntando quais alvos, em que estado, em que tecido, em que concentração e com que viés.
Por que alvos fora de CB1/CB2 importam clinicamente
É aqui que a ciência deixa de ser semântica e passa a afetar a medicina.
Para dor, alvos fora de CB1 podem ser a rota mais plausível para fármacos úteis com menos intoxicação. TRPV1, TRPA1, canais de sódio periféricos e vias transcricionais inflamatórias oferecem modos de reduzir a descarga de nociceptores ou a sensibilização neuroimune sem ativação central forte de CB1. Um relatório de 2026 da ScienceDaily sobre um composto de cannabis que “relieves pain without the high” é apenas um sinal em estágio de pesquisa, não uma resposta clínica final, mas a direção faz sentido. Se a analgesia puder ser deslocada para canais iônicos periféricos ou para exposição tecidual restrita, o antigo trade-off entre alívio da dor e carga psicoativa pode ser atenuado.
Para inflamação e metabolismo, PPAR-gamma é um bom exemplo de por que categorias de receptor importam. PPARs são receptores nucleares, não receptores cannabinoid de membrana. A ativação altera programas de expressão gênica envolvidos em manejo lipídico, sensibilidade à insulina e tônus inflamatório. Alguns efeitos de cannabinoid relatados em modelos metabólicos ou inflamatórios se encaixam melhor nessa biologia transcricional lenta do que na sinalização rápida de CB1. Mas, novamente, concentração e acesso intracelular importam. Um artigo que mostra ativação de PPAR em um ensaio com repórter não prova um efeito anti-inflamatório clinicamente relevante em humanos.[4]MIRA Pharmaceuticals Reports New Preclinical Data Demonstrating MIRA-55's Differentiated Mechanism of Action and Anxiolytic Activity Relative to THC. MIRA Pharmaceuticals. Nasdaq press release, 2025. https://www.nasdaq.com/press-release/mira-pharmaceuticals-reports-new-preclinical-data-demonstrating-mira-55s
Para ansiedade e náusea, mecanismos ligados à serotonina continuam reaparecendo, especialmente 5-HT1A. Os dados são mistos e, muitas vezes, indiretos, mas a persistência do sinal é reveladora. A reputação ansiolítica do CBD é difícil de mapear apenas para CB1/CB2. Essa é uma das razões pelas quais empresas estão tentando criar compostos inspirados em cannabinoid e diferenciados, em vez de simplesmente produzir análogos mais fortes de THC. Em 2025, a MIRA Pharmaceuticals relatou dados pré-clínicos alegando que seu candidato MIRA-55 mostrou um “differentiated mechanism of action” e “anxiolytic activity relative to THC.” Comunicados de imprensa de empresas são evidência de baixo nível e devem ser tratados assim. Ainda assim, revelam para onde o desenvolvimento de fármacos está caminhando: para longe da ideia de que o melhor medicamento cannabinoid é apenas uma estimulação mais limpa de CB1.
Prurido, enxaqueca, epilepsia, distúrbios intestinais e neuroproteção estão todos na mesma zona mecanística. Canais TRP regulam o ganho sensorial. GPRs podem moldar sinalização imune e epitelial. PPARs alteram programas inflamatórios. Canais de sódio controlam diretamente a excitabilidade. Vias de serotonina influenciam ansiedade, emese e respostas ao estresse. Uma vez que esses sistemas são colocados ao lado de CB1 e CB2, e não abaixo deles, muitos efeitos reais dos cannabinoid no mundo passam a parecer menos misteriosos e mais farmacologicamente comuns.
O modelo simplificado sobrevive porque é fácil. O modelo melhor sobrevive ao contato com os dados.
O sistema endocannabinoid versus o panorama mais amplo de alvos dos cannabinoid
A escrita popular sobre cannabis muitas vezes trata a farmacologia como uma história de dois receptores: CB1 explica os efeitos psicoativos, CB2 explica os efeitos imunológicos, e todo o resto é detalhe. Esse enquadramento é pequeno demais para as evidências. Ele ignora por que o cannabidiol não pode ser explicado de forma limpa por CB1 ou CB2, por que alguns cannabinoid provocam ardor ou analgesia por meio de canais TRP, por que receptores nucleares intracelulares como PPAR-gamma continuam aparecendo em estudos de inflamação e por que até o próprio THC pode afetar canais de sódio relevantes para a dor fora da sinalização cannabinoid clássica. Se o campo quiser explicar dor, ansiedade, inflamação, controle de crises ou problemas de segurança com intoxicantes novos, o reducionismo de receptor precisa acabar.
O momento regulatório deixa isso claro. Em 2025, o HHS afirmou que “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” ao apoiar a ação de agendamento contra produtos de 7-OH intensificado. A declaração não era sobre cannabis, mas captura a mesma lição farmacológica: quando os fabricantes passam de constituintes vegetais familiares para intoxicantes intensificados, semissintéticos ou estruturalmente modificados, os rótulos simples de categoria deixam de ser úteis. “THC-like” diz muito menos do que perfil-alvo, potência, metabólitos, distribuição tecidual e atividade fora do alvo.
Alvos canônicos: CB1, CB2, anandamida e 2-AG
O sistema endocannabinoid canônico continua importante. CB1 e CB2 são receptores acoplados à proteína G, principalmente acoplados a Gi/o, identificados no fim do século 20 e mapeados em detalhes por pesquisadores como Ken Mackie e Vincenzo Di Marzo. CB1 é fortemente expresso no sistema nervoso central, especialmente no córtex, hipocampo, gânglios da base e cerebelo, motivo pelo qual o agonismo parcial de THC ali se liga à intoxicação, aos efeitos sobre a memória, ao controle motor alterado e às mudanças de apetite. CB2 é enriquecido em células imunes e tecidos periféricos, embora não esteja ausente do cérebro. A ativação de qualquer um dos receptores geralmente reduz a formação de cAMP, modula canais iônicos e altera a liberação de neurotransmissores.
Os ligantes endógenos são anandamida e 2-arachidonoylglycerol, geralmente abreviado como anandamida e 2-AG. O grupo de Raphael Mechoulam foi central nessa história: a anandamida foi identificada em 1992, e o 2-AG, pouco depois. Eles não são armazenados em vesículas sinápticas como neurotransmissores clássicos. São sintetizados sob demanda a partir de precursores lipídicos de membrana e frequentemente atuam de modo retrógrado, movendo-se de células pós-sinápticas de volta aos terminais pré-sinápticos para atenuar a liberação de neurotransmissores. A anandamida é degradada principalmente pela FAAH; o 2-AG, principalmente pela MAGL. Esse ciclo bioquímico é a espinha dorsal do sistema endocannabinoid.
Mas a espinha dorsal não é o esqueleto inteiro. A anandamida também é agonista de TRPV1. CBD tem baixa afinidade direta por CB1 e CB2 em comparação com THC, mas claramente possui ações clinicamente significativas; a solução oral de cannabidiol aprovada pela FDA é indicada para convulsões associadas à síndrome de Lennox-Gastaut, à síndrome de Dravet e ao complexo de esclerose tuberosa em pacientes com 1 ano ou mais. Esse uso aprovado é um lembrete permanente de que efeitos cannabinoid clinicamente relevantes não precisam se alinhar com forte agonismo de CB1.
O que conta como alvo cannabinoid
Uma definição prática é melhor do que uma purista. Um alvo cannabinoid é qualquer sítio molecular no qual um phytocannabinoid, endocannabinoid, metabólito ou estrutura inspirada em cannabinoid se liga ou modula funcionalmente a sinalização em concentrações que podem importar em células, tecidos, animais ou humanos. Por esse padrão, o panorama se amplia rapidamente.
Os canais TRP são os exemplos não-CB mais familiares. TRPV1, TRPA1, TRPV2 e TRPM8 reaparecem em artigos sobre cannabinoid. Isso não é um detalhe lateral. David Julius e Ardem Patapoutian dividiram o Prêmio Nobel de Fisiologia ou Medicina de 2021 “for their discoveries of receptors for temperature and touch”, lembrando que os canais iônicos que governam calor, frio, irritação e mecanossensação estão diretamente nas vias da dor. A anandamida ativa TRPV1. CBD, CBG, CBC e cannabinoid ácidos mostraram atividade em TRP in vitro, muitas vezes com efeitos sensíveis à concentração e, por vezes, bifásicos. Um cannabinoid que primeiro ativa TRPV1 pode depois dessensibilizá-lo, produzindo o paradoxo de irritação inicial seguida de analgesia.
PPARs ampliam ainda mais o quadro. PPAR-α e PPAR-gamma são receptores nucleares que regulam transcrição relacionada ao metabolismo lipídico e à inflamação. Alguns cannabinoid e lipídios relacionados ao endocannabinoid atuam aqui diretamente ou após acúmulo intracelular e metabolismo. Esses são efeitos transcricionais mais lentos, não a sinalização em milissegundos de CB1. Isso importa para alegações de inflamação crônica, que muitas vezes fazem mais sentido por meio de sinalização nuclear do que por atividade aguda de receptores cannabinoid sinápticos.
Depois vêm os GPCR órfãos ou ainda debatidos, especialmente GPR55, GPR18 e GPR119. GPR55 foi repetidamente proposto como candidato a “CB3”, e o rótulo continua prematuro. O receptor é real; a classificação é debatida. CBD costuma ser descrito como antagonista ou modulador negativo de GPR55 em sistemas experimentais, enquanto certos lipídios endógenos e ligantes sintéticos podem ativá-lo. GPR18 e GPR119 aparecem em inflamação, metabolismo e sinalização imune, mas as evidências são irregulares e os efeitos entre espécies podem ser substanciais.
Receptores de serotonina, especialmente 5-HT1A, também pertencem a esse mapa mais amplo. A literatura sobre efeitos ansiolíticos e antieméticos do CBD frequentemente implica 5-HT1A, embora ainda se discuta se isso ocorre por agonismo direto ou facilitação indireta. Essa distinção importa. Um composto que se liga fracamente a um receptor, mas desloca de forma confiável o comportamento de circuito por mecanismos alostéricos ou em rede, ainda pode ter efeitos relevantes in vivo. A mesma cautela se aplica a programas pré-clínicos reportados por empresas: em 2025, a MIRA Pharmaceuticals disse que seu candidato MIRA-55 tinha um “differentiated mechanism of action” e apresentava atividade ansiolítica em relação ao THC. Isso não confirma benefício clínico, mas mostra para onde a química medicinal está indo — para longe de uma imitação grosseira de THC e em direção a uma farmacologia cannabinoid moldada pelo alvo.[5]Psychoactive cannabinoid THC inhibits peripheral nociceptors by targeting NaV1.7 and NaV1.8 nociceptive sodium channels. Hebrew University of Jerusalem cannabinoids research portal. Research portal summary, 2025. https://cannabinoids.huji.ac.il/publications/psychoactive-cannabinoid-thc-inhibits-peripheral-nociceptors-targeting[6]A cannabis compound that relieves pain without the high. ScienceDaily. ScienceDaily, 2026. https://www.sciencedaily.com/releases/2026/06/260619033343.htm
Os canais de sódio também merecem lugar aqui. Um relatório de 2025 da Hebrew University identificou a inibição de nociceptores periféricos pelo THC por meio de canais de sódio nociceptivos NaV1.7 e NaV1.8. Esse é um achado sério porque NaV1.7 e NaV1.8 são alvos centrais na dor, e o mecanismo fica fora de CB1/CB2. Também se alinha a uma mudança translacional maior. Em 2026, a ScienceDaily destacou pesquisas sobre “a cannabis compound that relieves pain without the high.” O composto exato e as perspectivas clínicas exigem escrutínio cuidadoso, mas a direção é crível: a analgesia pode, em princípio, ser separada da intoxicação central ao atingir canais iônicos periféricos ou alvos fora de CB, ou ambos.
Signaling bias A property of a ligand in which it stabilizes receptor states that favor one downstream pathway over another, such as G protein signaling over beta-arrestin recruitment.
Afinidade, eficácia, viés e janelas de concentração
Esse mapa mais amplo de alvos só faz sentido se os termos farmacológicos estiverem claros. Ki é uma constante de afinidade de ligação: um Ki menor geralmente significa ligação mais forte em um ensaio de competição. EC50 é a concentração que produz 50% de um efeito funcional medido. Eles não são intercambiáveis. Um ligante pode se ligar firmemente e ainda produzir sinalização fraca, ou ligar-se de forma moderada e, ainda assim, deslocar fortemente a função por amplificação na via.
Um agonista ativa um receptor. Um antagonista bloqueia a ativação por outro ligante. Um agonista inverso empurra receptores com atividade constitutiva para uma sinalização basal mais baixa. THC em CB1 costuma ser descrito como um agonista parcial: mesmo quando ocupa os receptores, não produz o efeito pleno de um agonista de alta eficácia. Isso ajuda a explicar por que diferentes cannabinoid — e até diferentes ligantes sintéticos de CB1 — podem ter tetos fisiológicos muito distintos.
Viés de sinalização significa que um ligante estabiliza conformações do receptor que favorecem uma via em detrimento de outra, como sinalização por proteína G em vez de recrutamento de β-arrestina. Esse já é um pensamento padrão em desenvolvimento de fármacos, inclusive na química medicinal de cannabinoid; o artigo de 2016 no Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, está nessa tradição orientada por alvo. Dessensibilização significa que ativação repetida ou sustentada pode reduzir a responsividade, um problema importante para canais TRP e para o próprio CB1. Por fim, engajamento de alvo específico de tecido significa que o mesmo composto pode atingir alvos diferentes no cérebro, intestino, pele, células imunes ou nervos periféricos, dependendo da concentração, via, metabolismo e expressão local de proteínas. É por isso que a promiscuidade in vitro não equivale automaticamente à relevância clínica — mas também por que explicações apenas em CB1/CB2 continuam falhando.
Canais TRP: os sensores de calor, dor e irritação que os cannabinoid continuam atingindo
O resumo usual diz que os cannabinoid atuam por meio de CB1 e CB2. Isso é estreito demais para explicar o que muitas dessas moléculas realmente fazem no tecido. Repetidamente, phytocannabinoid atingem canais de potencial receptor transitório, uma superfamília de canais iônicos presente em nociceptores, queratinócitos, nervos respiratórios, células imunes e outras interfaces sensoriais onde o corpo detecta calor, frio, substâncias químicas, estiramento, lesão e inflamação.[7]The Nobel Prize in Physiology or Medicine 2021. The Nobel Assembly at Karolinska Institutet. Nobel Prize Press Release, 2021. https://www.nobelprize.org/prizes/medicine/2021/press-release/
Essa biologia não é obscura. Ela foi suficientemente central para a ciência somatossensorial a ponto de o Prêmio Nobel de Fisiologia ou Medicina de 2021 ter ido para David Julius e Ardem Patapoutian “for their discoveries of receptors for temperature and touch.” O trabalho de Julius identificando o receptor da capsaicina, TRPV1, ajudou a consolidar a visão moderna de que a sinalização da dor não é apenas um fio que carrega informações de dano; ela é quimicamente regulada já na primeira terminação sensorial. Isso importa para os cannabinoid porque vários cannabinoid vegetais interagem com a mesma maquinaria molecular que responde à pimenta, ao óleo de mostarda, ao calor nocivo, a agentes refrescantes, a condições ácidas e a lipídios inflamatórios.
O resultado é uma farmacologia que parece confusa se você espera um receptor e um efeito. Faz mais sentido se você pensar em controle de ganho sensorial. Muitos cannabinoid são ligantes de baixa a moderada afinidade para receptores CB e, ao mesmo tempo, moduladores diretos de canais TRP. Alguns os ativam. Outros os inibem. Alguns fazem ambos, dependendo da concentração, da espécie, da variante de splicing, do ambiente de membrana e de o ensaio medir influxo de cálcio, corrente, liberação de neuropeptídeos ou comportamento em um animal.
TRPV1, TRPA1, TRPV2 e TRPM8 na biologia sensorial
Os canais TRP são detectores polimodais. TRPV1 é o mais conhecido: ativado por capsaicina, calor nocivo, prótons e mediadores inflamatórios endógenos, ele é fortemente expresso em neurônios sensoriais de pequeno diâmetro que dirigem dor em queimação e inflamação neurogênica. Abra o canal e cátions entram, despolarizando o neurônio e aumentando o cálcio intracelular. TRPA1 costuma estar em populações sobrepostas de nociceptores e é famoso por detectar irritantes eletrofílicos, como isotiocianato de alila da mostarda e do wasabi, acroleína da fumaça e produtos de estresse oxidativo gerados durante a inflamação. Ele é relevante não apenas para dor, mas também para prurido, tosse, hiperreatividade das vias aéreas e sinalização trigeminal semelhante à da enxaqueca.
TRPV2 é menos direto. É um canal termo- e mecanossensível de limiar alto em alguns sistemas, mas também aparece em células imunes, glia e tecidos proliferativos, razão pela qual continua surgindo em discussões sobre inflamação e, mais especulativamente, biologia do câncer. TRPM8, por outro lado, é o sensor clássico de frio, ativado por baixas temperaturas e compostos como mentol e icilina. Ainda assim, ele também importa em estados de dor, nos quais a alodinia ao frio pode se tornar severa, e, em alguns contextos, a atividade de TRPM8 pode suprimir a dor por contrarritmo em nível de circuito. Mesma família, papéis sensoriais muito diferentes.
Esse espalhamento de funções explica por que os efeitos dos cannabinoid podem parecer contraditórios à primeira vista. Ativar TRPV1 ou TRPA1 pode arder. Bloquear TRPM8 pode reduzir a sensação de frio, mas também alterar a dor ao frio. Estimular TRPV2 em um tipo celular pode afetar a sinalização de cálcio sem produzir qualquer efeito sensorial óbvio. Não existe um único “efeito TRP”, assim como não existe um único “efeito cannabinoid”.
CBD, CBG, CBC e THC em canais da família TRP
Entre os phytocannabinoid, o CBD tem o perfil TRP mais forte e mais reproduzido. Em sistemas de expressão heteróloga, o CBD ativa TRPV1, TRPA1 e TRPV2 humanos em concentrações micromolares e inibe TRPM8. Um estudo amplamente citado de De Petrocellis e colegas, de 2011, usando imagem de cálcio em células HEK-293 transfectadas, concluiu que o CBD se comportava como agonista em TRPV1, TRPV2, TRPA1 e TRPV4, ao mesmo tempo em que antagonizava TRPM8. A potência não era uniforme: TRPA1 era especialmente sensível, com atividade em baixa faixa micromolar, enquanto outros canais exigiam concentrações um pouco mais altas. Esse padrão se sustentou o suficiente para que o engajamento de TRP agora faça parte de qualquer relato sério sobre a farmacologia do CBD.
CBG e CBC se encaixam no mesmo tema geral, embora com impressões próprias. O CBG mostrou repetidamente atividade em TRPA1 e TRPV1, além de inibição de TRPM8, o que o torna farmacologicamente interessante para modelos de dor inflamatória e hipersensibilidade visceral. CBC é menos estudado que CBD, mas trabalhos in vitro disponíveis sugerem que ele também ativa TRPA1 e pode envolver TRPV1. Não se trata de pequenas curiosidades vistas em um único ensaio e nunca mais. Elas reaparecem em sistemas recombinantes e em preparações sensoriais primárias, exatamente por isso retornam em artigos mecanísticos sobre analgesia e inflamação.
THC é mais complicado. Pode ativar TRPV2 e já foi relatado como interagindo com TRPA1 e TRPV1 em algumas condições, mas sua farmacologia é dominada em muitos experimentos por efeitos mediados por CB1, especialmente no sistema nervoso central. Ainda assim, a ideia de que THC é apenas um fármaco de CB1 está errada. Trabalhos recentes da Hebrew University, relatados em 2025, argumentaram que THC inibe nociceptores periféricos ao atingir canais de sódio NaV1.7 e NaV1.8, um mecanismo não-CB separado que reforça o ponto geral: os cannabinoid frequentemente atingem múltiplos alvos relevantes para dor ao mesmo tempo. Canais TRP fazem parte desse mapa mais amplo fora de CB.
É necessária uma ressalva. Grande parte dessas evidências vem de ensaios em faixa micromolar, e nem toda micromolar em uma placa corresponde a uma concentração livre alcançável no sítio-alvo humano. Cannabinoid lipofílicos se particionam em membranas, ligam-se a proteínas e geram metabólitos; a via de administração e o acúmulo no tecido importam. O fato de a solução oral de CBD ser aprovada pela FDA para distúrbios convulsivos não prova que TRPV1 ou TRPA1 sejam os motores de seus efeitos clínicos em epilepsia. Apenas mostra que o CBD claramente faz coisas em humanos que não podem ser capturadas chamando-o de “composto cannabinoid de receptor CB não intoxicante”. A história molecular é maior do que esse rótulo.
A atividade em TRP também é sensível ao ensaio. Um canal pode parecer “ativado” em um ensaio de cálcio porque estoques intracelulares, potencial de membrana ou lipídios endógenos estão mudando em paralelo. Diferenças de espécie podem ser reais. O mesmo vale para dependência do estado. Tecidos inflamados acidificam, oxidam e produzem mediadores lipídicos, todos os quais remodelam o gating de TRP. Um cannabinoid que mal move um canal no estado basal pode ter um efeito muito maior em uma terminação nervosa lesionada.
Dessensibilização, analgesia e por que ativação pode reduzir a dor
Esta é a parte que confunde não especialistas: se TRPV1 e TRPA1 são canais que produzem dor, por que ativá-los jamais reduziria a dor?
Porque ativação aguda e saída funcional sustentada não são a mesma coisa.
TRPV1 é o exemplo clássico. A capsaicina inicialmente arde, depois dessensibiliza nociceptores e pode produzir analgesia após exposição repetida ou em alta concentração. Clinicamente, esse princípio é usado no adesivo de capsaicina a 8% para dor neuropática. O mecanismo inclui dessensibilização dependente de cálcio, depleção de neuropeptídeos como substância P e CGRP, alteração do estado de fosforilação do canal e, em alguns casos, desfuncionalização reversível da terminação nervosa. Um canal que dispara fortemente no início pode tornar-se menos responsivo depois. O sinal imediato é pró-nociceptivo; o estado subsequente pode ser anti-nociceptivo.
Os cannabinoid parecem explorar a mesma lógica. A ativação de TRPV1 ou TRPA1 pelo CBD pode desencadear entrada de cálcio e, em seguida, reduzir a responsividade do canal e amortecer a excitabilidade dos neurônios sensoriais. Essa é uma via plausível pela qual um composto pode arder em uma placa de Petri, mas reduzir hiperalgesia em um animal. O eixo temporal importa. A dose também. Baixas concentrações podem sensibilizar ou ativar fracamente. Concentrações mais altas podem levar à dessensibilização ou até a efeitos mais amplos de membrana que suprimem a descarga.
TRPA1 adiciona outra camada porque está profundamente ligado a irritantes inflamatórios e estresse oxidativo. Em sistemas de vias aéreas e trigeminais, a ativação repetida ou prolongada pode alterar a liberação de neuropeptídeos e a responsividade reflexa. Isso o torna relevante para tosse, enxaqueca e estados de exacerbação inflamatória, e não apenas para “dor” em sentido estrito. Se um cannabinoid se liga a TRPA1 e depois reduz a responsividade subsequente, o efeito líquido pode ser menos sinalização de irritação, mesmo que o primeiro evento molecular tenha sido a abertura do canal.
TRPM8 mostra o tipo oposto de padrão cannabinoid em muitos ensaios: cannabinoid como CBD e CBG frequentemente o inibem, em vez de ativá-lo. Isso pode importar em hipersensibilidade ao frio, em que a sinalização excessiva de TRPM8 contribui para alodinia dolorosa ao frio. Aqui não há o paradoxo de ativação levando ao alívio; a hipótese mais simples é a supressão direta de uma via sensível ao frio. Mas isso também não deve ser superestimado. Em alguns estados de dor, a atividade de TRPM8 pode contrariar a dor ao calor ou o prurido, então bloqueá-lo não é automaticamente benéfico.
A posição mais forte que as evidências sustentam é esta: os canais TRP não são notas de rodapé na farmacologia dos cannabinoid. São alvos recorrentes e funcionalmente relevantes, especialmente para efeitos sensoriais periféricos envolvendo calor, irritação química, dor inflamatória, prurido e reflexos das vias aéreas. Eles não explicam tudo. Nem sempre são o mecanismo dominante in vivo. Ainda assim, qualquer pessoa tentando entender por que CBD, CBG, CBC ou até THC podem alterar dor e inflamação sem correspondência limpa com CB1 ou CB2 precisa de TRPV1, TRPA1, TRPV2 e TRPM8 na página cedo, e não como um pensamento tardio.
Isso também importa para o desenvolvimento de fármacos. Agências de saúde pública já estão distinguindo cannabinoid familiares de intoxicantes quimicamente alterados ou intensificados, porque diferenças no nível do alvo podem mudar o risco. O mesmo princípio vale ao contrário para terapêuticos: se a analgesia puder ser separada da intoxicação central, uma rota é desenhar compostos que enviesem a ação para canais TRP periféricos e outros alvos fora de CB, em vez de agonismo forte de CB1 com penetração cerebral. A velha história reducionista de receptor é pequena demais para os dados.
PPARs: cannabinoid como sinais lipídicos intracelulares, e não apenas ligantes de receptores de membrana
Os receptores ativados por proliferadores de peroxissoma, geralmente abreviados como PPARs, mudam a conversa sobre cannabinoid porque ficam em um lugar diferente e operam em um relógio diferente de CB1 e CB2. CB1 e CB2 são receptores de membrana acoplados à proteína G, feitos para sinalização rápida: segundos a minutos, canais iônicos, liberação de neurotransmissores, cascatas de quinases. PPARs são receptores nucleares. Eles respondem a moléculas lipofílicas, acionam maquinarias transcricionais e remodelam quais genes uma célula expressa ao longo de horas a dias. Essa mudança importa. Significa que alguns efeitos de cannabinoid podem parecer menos com agonismo clássico de receptor e mais com reprogramação regulada por lipídios do tônus inflamatório, do manejo mitocondrial, da oxidação de ácidos graxos, da sinalização fibrótica e das respostas gliais.
Isso não é um salto especulativo. Os cannabinoid são altamente lipofílicos, acumulam-se em membranas, particionam-se em compartimentos intracelulares e geram metabólitos que podem ter perfis-alvo diferentes da molécula original. Uma classe de fármacos com essas propriedades quase parece feita para continuar esbarrando em sensores lipídicos nucleares. PPARs estão entre os lugares mais plausíveis para isso ocorrer.
O que PPAR-alpha, PPAR-gamma e PPAR-delta fazem
Os três principais isoformas de PPAR se sobrepõem, mas não são intercambiáveis. PPAR-alpha está classicamente ligado ao catabolismo de ácidos graxos. É abundante em fígado, coração, rim, músculo e outros tecidos que queimam gordura de forma agressiva e, quando ativado, impulsiona programas transcricionais de beta-oxidação, cetogênese, manejo de lipoproteínas e menor sinalização inflamatória. Farmacologistas o conhecem por meio dos fibratos. Em pesquisa sobre dor e inflamação, PPAR-alpha também importa fora do metabolismo porque pode suprimir a expressão de genes inflamatórios ligados ao NF-kappaB e alterar a sinalização sensorial.
PPAR-gamma é o isoforma que continua aparecendo em artigos sobre cannabinoid, às vezes por bons motivos e às vezes porque é a história mais fácil de contar. Ele é muito relevante para diferenciação de adipócitos e sensibilidade à insulina, mas esse resumo o subestima. PPAR-gamma regula polarização de macrófagos, produção de citocinas, respostas ao estresse oxidativo, remodelação fibrótica, comportamento endotelial e ativação glial no sistema nervoso central. Isso lhe dá relevância óbvia para doença inflamatória intestinal, neuroinflamação, complicações diabéticas e fibrose tecidual. Também é um alvo de dupla face: ativação forte pode melhorar a sensibilidade à insulina, mas trazer edema, ganho de peso e outras limitações familiares dos tiazolidinedionas.
PPAR-delta, também chamado PPAR-beta/delta, recebe menos atenção na escrita popular sobre cannabinoid, mas não deveria. É amplamente expresso e sustenta o uso de ácidos graxos, a função mitocondrial, a reparação de feridas, a biologia dos queratinócitos e alguns programas anti-inflamatórios. Dependendo do contexto, pode tanto conter quanto facilitar processos de doença, o que explica por que a literatura sobre ele é menos organizada. Se um cannabinoid ou metabólito de cannabinoid envolve PPAR-delta, a leitura biológica pode variar muito mais por tecido do que uma história simples de “agonista=benefício” sugere.
Mecanisticamente, todos os três isoformas funcionam como fatores de transcrição ativados por ligante que heterodimerizam com o receptor X de retinoide e se ligam a elementos de resposta proliferadora de peroxissoma no DNA. Uma vez engajados, eles não ligam apenas um interruptor. Alteram redes transcricionais. Coativadores, corepressores, estado da cromatina, tipo celular, contexto inflamatório e conformação específica do receptor para aquele ligante tudo influencia o resultado. Dois compostos podem ambos ser chamados de agonistas de PPAR-gamma e ainda assim conduzir biologia significativamente diferente.
Esse ponto é especialmente importante para cannabinoid, que muitas vezes são moléculas farmacologicamente promíscuas, em vez de ferramentas limpas de alvo único.
CBD e cannabinoid relacionados na sinalização metabólica e inflamatória
CBD é o exemplo recorrente porque seu perfil clínico é mal explicado apenas por CB1 ou CB2. A solução oral aprovada pela FDA para crises associadas à síndrome de Lennox-Gastaut, à síndrome de Dravet e ao complexo de esclerose tuberosa mostra que o CBD é farmacologicamente real em humanos, mas não que algum único alvo fora dos cannabinoid explique suas ações. PPAR-gamma é um dos candidatos mais citados porque diversos estudos celulares e animais relacionaram o CBD a efeitos anti-inflamatórios e metabólicos atenuados por antagonistas de PPAR-gamma ou acompanhados por mudanças transcricionais dependentes de PPAR-gamma.
Um artigo amplamente citado de O’Sullivan e colegas, de 2009, relatou que o CBD causava vasorrelaxamento em artérias humanas e que parte do efeito era sensível ao antagonista de PPAR-gamma GW9662, sugerindo um componente dependente de PPAR-gamma. Em 2011, Esposito e coautores mostraram, em um modelo celular semelhante ao de Alzheimer, que o CBD reduziu a neuroinflamação induzida por beta-amiloide e que o bloqueio de PPAR-gamma reduziu esse efeito protetor. Em 2013, Hind e O’Sullivan revisaram evidências de que cannabinoid podem ativar PPARs diretamente ou indiretamente, enquadrando CBD, THC, ácido ajulemico, lipídios relacionados à anandamida e vários cannabinoid sintéticos.
O padrão é consistente o suficiente para levar a sério: o CBD frequentemente cai em sistemas experimentais nos quais genes inflamatórios diminuem, marcadores de estresse oxidativo caem e o antagonismo de PPAR-gamma enfraquece a resposta. Mas levar a sério não é o mesmo que considerar isso resolvido. Muitos desses estudos usam concentrações micromolares de CBD. Isso importa porque as concentrações livres intracelulares em tecidos humanos vivos são difíceis de inferir a partir de concentrações nominais de banho em uma placa. O CBD também se liga a membranas e as perturba, afeta o manejo de cálcio, interage com canais TRP, influencia a sinalização da adenosina ao inibir o transporte de nucleosídeos e pode alterar o tônus endocannabinoid. Qualquer uma dessas rotas pode alimentar mudanças transcricionais que depois parecem “tipo PPAR”.
Cannabinoid relacionados reforçam o caso sem resolvê-lo. THC foi relatado em alguns sistemas como ativador de PPAR-gamma, embora geralmente de forma fraca em comparação com ligantes dedicados. Ácido cannabidiólico e ácido tetraidrocanabinólico mostraram atividade de PPAR em ensaios selecionados. Lipídios relacionados ao endocannabinoid, como palmitoiletanolamida, oleoiletanolamida e alguns derivados oxidados, têm relações mais fortes e mais estabelecidas com PPAR-alpha e PPAR-gamma do que os phytocannabinoid mais conhecidos. Essa é uma das razões pelas quais o enquadramento da sinalização lipídica intracelular é melhor do que o enquadramento estreito “cannabinoid vegetais se ligam a PPARs”. A espécie ativa pode ser o cannabinoid original, um metabólito, um mediador lipídico coadministrado ou uma mudança subsequente nos pools lipídicos endógenos.
O ácido ajulêmico é um caso útil. É um análogo sintético relacionado ao THC, mas desenvolvido intencionalmente para se afastar da intoxicação clássica. Em trabalhos pré-clínicos, mostrou ações anti-inflamatórias e antifibróticas com evidências implicando PPAR-gamma entre outros alvos. Esse tipo de química medicinal espelha uma tendência mais ampla no campo. Já em 2016, um artigo do ACS Journal of Medicinal Chemistry intitulado “Library Docking for Cannabinoid-2 Receptor Ligands” refletia o design estruturado por alvo em vez de rótulos grosseiros de receptor, e programas mais recentes de cannabinoid buscam cada vez mais separar analgesia, ansiolise ou imunomodulação da ativação central de CB1. A mesma lógica vale para estruturas ativas em PPAR: se uma biologia cannabinoid útil pode ser extraída por mecanismos transcricionais e periféricos, não há razão para que o desenvolvimento de fármacos permaneça preso à farmacologia tipo THC.
Os dados sobre sinalização metabólica do CBD são mais mistos do que os dados anti-inflamatórios. Alguns estudos pré-clínicos sugerem melhora da sensibilidade à insulina, redução de adipocinas inflamatórias ou melhor manejo mitocondrial. Outros não mostram benefício importante, e as evidências em humanos são frágeis. A discussão pública muitas vezes se antecipa demais aos dados aqui. O fato de PPAR-gamma controlar a biologia da glicose e do tecido adiposo não significa que o CBD seja um modulador metabólico clinicamente significativo em humanos nas exposições padrão.
Transcrição gênica, efeitos tardios e limites da evidência
A biologia de PPAR exige uma correção no tempo. Se um efeito de cannabinoid aparece em segundos ou poucos minutos, PPARs provavelmente não são a explicação principal. A sinalização por receptores nucleares geralmente exige acesso do ligante a compartimentos intracelulares, engajamento do receptor, recrutamento alterado de co-reguladores, mudanças transcricionais e, então, consequências em nível proteico. Isso leva tempo. Horas são plausíveis. Dias são comuns. Quando artigos afirmam que o efeito rápido de um cannabinoid é “via PPAR-gamma”, o ceticismo é apropriado, a menos que o desenho separe claramente a sinalização não genômica imediata dos desfechos transcricionais tardios.
O problema recorrente é o desenho do ensaio. Ensaios com repórter podem mostrar que um composto aumenta a transcrição dependente de PPAR, mas sistemas repórter são artificiais e podem exagerar atividade fraca. Estudos com antagonistas são informativos, mas fármacos como GW9662 não são um soro da verdade mágico; efeitos fora do alvo e bloqueio parcial complicam a interpretação. Ensaios de ligação ajudam, mas a ligação direta não garante que a exposição tecidual alcance a concentração necessária in vivo. Modelos de knock-out são mais fortes, embora compensações por outras vias possam obscurecer os resultados. A melhor evidência combina métodos: engajamento direto do alvo, farmacologia seletiva para o receptor, perturbação genética, concentrações relevantes no tecido e uma curva temporal consistente com ação transcricional. Grande parte da literatura sobre cannabinoid-PPAR não alcança esse padrão.
A proeminência de PPAR-gamma na pesquisa com CBD é, portanto, tanto justificada quanto exagerada. Justificada, porque o sinal reaparece em modelos vasculares, inflamatórios, neurodegenerativos e relacionados à fibrose. Exagerada, porque o CBD é exatamente o tipo de molécula lipofílica, de múltiplos alvos, para a qual concentração intracelular, metabólitos ativos e contexto do ensaio podem criar histórias mecanísticas sedutoras, porém incompletas. Uma queda de TNF-alpha ou IL-6 após exposição ao CBD não é uma impressão digital. É uma pista.
Ainda assim, o ponto mais amplo permanece. Os cannabinoid não devem ser tratados apenas como ligantes de receptores cannabinoid de membrana. Alguns atuam, direta ou indiretamente, como sinais lipídicos intracelulares capazes de envolver maquinarias transcricionais nucleares. Isso abre rotas plausíveis para efeitos anti-inflamatórios, antifibróticos e neuroimunes mais lentos, menos ligados à intoxicação e potencialmente mais relevantes para modificação de doença de longo prazo do que a sinalização aguda de CB1. Também levanta uma lição regulatória. Como autoridades já enfatizaram em outros contextos, inclusive na declaração do HHS de 2025 de que produtos com 7-hydroxymitragynine intensificado representam “an imminent hazard to public safety”, diferenças em nível molecular importam. Pequenas mudanças estruturais podem redirecionar o engajamento de alvo. Para cannabinoid e produtos canabinoides, isso significa que a história de segurança e eficácia não pode ser inferida apenas pela familiaridade com THC, e a biologia de PPAR é uma das razões.
GPR55, GPR18 e GPR119 e o problema dos GPCR órfãos
Um GPCR órfão é um receptor acoplado à proteína G cujo ligante endógeno, papel fisiológico ou ambos permanecem incertos. Um receptor de órfão desanonimizado é aquele para o qual um ativador endógeno convincente foi proposto e replicado o suficiente para sustentar uma biologia de trabalho. Isso parece organizado. Na prática, raramente é. A farmacologia dos cannabinoid continua esbarrando nessa confusão porque endocannabinoid e phytocannabinoid são lipofílicos, ativos em membrana e promíscuos: podem alterar fluxo de cálcio, atividade de quinases ou transcrição de forma que parece mediada por receptor mesmo quando o alvo direto ainda não está resolvido. Foi exatamente assim que GPR55, GPR18 e GPR119 entraram na conversa como “nonclassical cannabinoid receptors”.
A tentação de cunhar um novo rótulo de receptor é forte. Gera manchetes. Também corre mais rápido que as evidências. GPR55 chegou mais perto de ser rotulado como “CB3”, mas o campo nunca alcançou a coerência que sustentou CB1 e CB2. O mesmo cuidado se aplica ainda mais a GPR18 e GPR119.
Por que GPR55 já foi chamado de possível receptor cannabinoid
GPR55 foi clonado em 1999, e levantamentos iniciais de expressão o colocaram em tecidos relevantes para a biologia cannabinoid: regiões do cérebro, gânglios da raiz dorsal, baço, trato gastrointestinal, vasculatura, células imunes e células relacionadas ao osso, incluindo osteoclastos e populações da linhagem de osteoblastos. Essa distribuição importava. Um receptor expresso em vias de dor, tecidos inflamatórios e osso convida imediatamente à comparação com CB1 e CB2, especialmente quando ligantes cannabinoid parecem alterar seus desfechos.
Seu perfil de sinalização também parecia suficientemente diferente para ser interessante. Ao contrário de CB1 e CB2, que se acoplam principalmente a Gi/o e tendem a inibir a adenilil ciclase, GPR55 sinaliza mais frequentemente por Gα12/13 e, às vezes, por vias ligadas a Gq, ativando RhoA, fosfolipase C, ERK e liberação de cálcio intracelular. Em ensaios celulares, o desfecho característico costuma ser um transiente de cálcio. Isso tornou GPR55 fácil de “ver” em sistemas heterólogos, mas também fácil de superestimar, porque ensaios de cálcio são sensíveis à densidade de receptor, ao background celular, à lipofilicidade do ligante e ao tempo do ensaio.
O motivo específico pelo qual GPR55 virou candidato a receptor cannabinoid foi o fato de vários cannabinoid e ligantes semelhantes a cannabinoid produzirem efeitos mensuráveis nele. Ryberg e colegas, escrevendo no British Journal of Pharmacology em 2007, relataram que GPR55 podia ser ativado por múltiplos ligantes cannabinoid e o propuseram como “a novel cannabinoid receptor.” Esse artigo se tornou a dobradiça histórica. Não resolveu a questão; criou-a.
Logo depois, as fissuras apareceram. Alguns grupos descobriram que lisofosfatidilinositol, especialmente espécies de 2-arachidonoil LPI, era um agonista endógeno mais convincente do que qualquer cannabinoid clássico. Oka e colegas, em 2007 e em trabalhos de seguimento posteriores, defenderam fortemente essa visão. Outros observaram que compostos frequentemente discutidos na pesquisa cannabinoid se comportavam de modo inconsistente em GPR55: cannabidiol (CBD) costumava parecer antagonista ou modulador negativo em alguns ensaios, enquanto Δ9-THC era fraco, parcial ou inativo, dependendo do sistema. Cannabidiol análogo, O-1602 e certos cannabinoid sintéticos às vezes mostravam atividade mais clara do que o próprio THC. Isso não é o que se espera de um terceiro receptor cannabinoid limpo.
Ainda assim, a biologia de GPR55 é real, mesmo que o rótulo seja instável. Na pesquisa sobre dor, o receptor é expresso em neurônios sensoriais e circuitos espinais, e a interrupção genética ou farmacológica da sinalização de GPR55 reduziu a hipersensibilidade mecânica em alguns modelos roedores. Staton e colegas, em Pain (2008), ligaram a ativação de GPR55 ao processamento da dor inflamatória e neuropática, com antagonismo reduzindo a hipersensibilidade. Porém, o efeito não é universal entre modelos ou ligantes. Alguns dados sugerem sinalização pronociceptiva por mobilização de cálcio e aumento da excitabilidade neuronal; outros conjuntos de dados são mais fracos ou limitados ao modelo. A leitura mais segura é que GPR55 pode contribuir para a sinalização de dor em alguns contextos, especialmente estados inflamatórios, mas não é um interruptor mestre da dor.
A biologia óssea oferece um sinal mais sólido. Por quê? Porque fenótipos de knock-out de GPR55 são mais difíceis de descartar como artefatos de ensaio. Em 2009, Whyte e colegas relataram no PNAS que camundongos sem GPR55 apresentavam maior massa óssea e função de osteoclasto prejudicada, argumentando que GPR55 promove a reabsorção por osteoclastos. Isso fazia sentido mecanístico com sua sinalização ligada a cálcio e RhoA. Osteoclastos dependem de reorganização do citoesqueleto e manejo localizado de cálcio; GPR55 se encaixa melhor nessa maquinaria do que CB1. Se um cannabinoid ou composto semelhante modula GPR55 aqui, a consequência fisiológica pode ser substancial.
Inflamação é o terceiro grande tema. GPR55 está presente em células relacionadas ao sistema imune, e sua ativação foi ligada à liberação de citocinas, ao comportamento de leucócitos e a respostas inflamatórias vasculares. Mas, novamente, a direção não é perfeitamente uniforme. Em algumas preparações, a ativação de GPR55 parece pró-inflamatória; em outras, mais regulatória, o que provavelmente reflete tipo celular, viés do ligante e comunicação cruzada entre receptores, e não uma contradição simples. Um receptor que se acopla por múltiplas vias e fica em ambientes de membrana diferentes não produzirá uma saída universal única.
Essa complexidade explica a disputa prolongada entre agonista e antagonista na literatura cannabinoid. CBD é o exemplo mais claro. Em vários estudos, o CBD se comportou como antagonista ou inibidor funcional de GPR55, atenuando a sinalização de cálcio dirigida por LPI. Lauckner e colegas, em 2008, em um artigo amplamente citado no PNAS, mostraram que a ativação de GPR55 aumentava o cálcio intracelular e promovia liberação de neurotransmissores, enquanto o CBD contrabalançava aspectos dessa sinalização. Isso alimentou uma hipótese persistente de que alguns efeitos do CBD, especialmente em modelos de convulsão e inflamação, podem envolver em parte o bloqueio de GPR55 e não ação em CB1 ou CB2. Essa ideia é plausível. Não está comprovada como mecanismo dominante em humanos.
THC é ainda mais confuso. Alguns relatos o classificam como agonista de baixa potência de GPR55; outros encontram eficácia negligenciável; outros sugerem comportamento que depende da reserva do receptor ou da via medida. Um ligante pode parecer agonista em um ensaio de β-arrestina, neutro em ligação e antagonista em um ensaio de cálcio se o sistema estiver superexpresso ou enviesado. Isso não é detalhe técnico. É a história.
A evidência mista para GPR18 e GPR119
GPR18 foi frequentemente discutido porque responde em alguns sistemas à N-arachidonoil glicina, um lipídio relacionado ao endocannabinoid, e porque cannabidiol análogo e compostos relacionados mostraram efeitos vasculares ou imunes que alguns autores mapearam para GPR18. A expressão foi relatada em células imunes, microglia, baço e alguns tecidos periféricos. Isso o tornou atraente como candidato à regulação inflamatória, tráfego imune e possivelmente dor.
Mas a farmacologia foi irregular desde o início. Kohno e colegas, em 2006, apoiaram a ativação de GPR18 por N-arachidonoil glicina. McHugh e colegas depois associaram GPR18 à migração microglial e à sinalização inflamatória. Então vieram os problemas de replicação. Alguns laboratórios não conseguiam reproduzir respostas ao ligante em sistemas transfectados. Outros encontravam forte dependência de marcação do receptor, da linhagem celular ou do ortólogo de espécie. Um receptor que “funciona” apenas em uma arquitetura de ensaio não está desanonimizado em qualquer sentido estável. Especificamente para cannabinoid, a evidência é mais fraca do que os resumos populares sugerem. Pode haver biologia real ali, mas o caso de GPR18 como um receptor cannabinoid genuíno ainda é frágil.
GPR119 é diferente. É muito menos plausível como receptor cannabinoid, apesar de às vezes ser incluído em listas amplas de receptores “fora de CB”. GPR119 está principalmente associado à detecção de lipídios em células beta pancreáticas e células enteroendócrinas, acoplando-se por Gs para elevar cAMP e promover secreção de insulina dependente de glicose e liberação de incretinas. Oleoiletanolamida é um candidato a ligante endógeno melhor estabelecido do que qualquer cannabinoid clássico. Como alguns etanolamidas de ácidos graxos são estruturalmente adjacentes à química do endocannabinoid, GPR119 pode ser arrastado para discussões sobre cannabinoid por associação. Isso é, em grande parte, confusão de categoria. A sobreposição é vizinhança química, não evidência forte de que THC, CBD ou principais phytocannabinoid ajam de forma relevante por meio de GPR119 em concentrações fisiológicas.
O que a farmacologia de receptores órfãos erra nas manchetes
A falha padrão da mídia é simples: um ensaio positivo de sinalização vira “cientistas descobriram um novo receptor cannabinoid.” Esse salto ignora pelo menos quatro filtros.
Primeiro, dependência do ensaio. Mobilização de cálcio, recrutamento de β-arrestina, fosforilação de ERK, redistribuição dinâmica de massa e ligação radioligante não fazem a mesma pergunta. Um ligante lipofílico pode perturbar membranas, alterar o tráfego do receptor ou mostrar viés de via. Se o receptor está superexpresso, compostos fracos passam a parecer fortes.
Segundo, diferenças entre espécies. GPR55 humano não é GPR55 de camundongo em todos os detalhes farmacológicos, e o mesmo vale para GPR18. Um perfil de ligante construído em HEK293 com o receptor humano pode não prever um estudo de dor em ratos.
Terceiro, concentração. Muitos artigos sobre cannabinoid relatam atividade micromolar in vitro. Isso pode importar farmacologicamente, mas não automaticamente. Níveis teciduais após inalação, dose oral, metabolismo de primeira passagem ou acúmulo local em gordura e membranas variam enormemente. Ligação in vitro não é mecanismo clínico.
Quarto, contexto. Um receptor em células imunes pode mediar um efeito; o mesmo receptor em osteoclastos, outro. Adicione comunicação cruzada com canais TRP, PPARs, receptores de serotonina e até canais de sódio, e a história limpa de um ligante/um receptor se desfaz rapidamente.
É por isso que “CB3” nunca pegou. GPR55 tem biologia crível em sinalização de cálcio, dor, remodelação óssea e inflamação. Também apresenta farmacologia cannabinoid contraditória, alta sensibilidade a ensaio e uma reivindicação concorrente forte de que lipídios da família LPI são seus ligantes fisiológicos primários. GPR18 é ainda mais incerto. GPR119, em grande parte, não pertence ao mesmo grupo, exceto como lembrete de que GPCR lipídicos são fáceis de associar em excesso aos cannabinoid.
Para a ciência dos cannabinoid, a lição é moderação. Esses receptores podem importar muito. Só não justificam renomeação prematura.
Sinalização de serotonina: onde os cannabinoid se cruzam com os sistemas 5-HT
A serotonina é o ponto em que muitas alegações populares sobre CBD se tornam ao mesmo tempo mais plausíveis e mais escorregadias. A parte plausível é simples: em ensaios celulares, modelos de ansiedade em roedores, paradigmas de estresse e um pequeno número de estudos experimentais em humanos, 5-HT1A continua aparecendo como um nó relevante nos efeitos comportamentais do CBD. A parte escorregadia é que “atua em serotonina” pode significar várias coisas diferentes. Pode significar agonismo direto no sítio ortostérico. Pode significar modulação alostérica positiva. Pode significar facilitação da sinalização do receptor sem ligação de alta afinidade. Ou pode significar que o CBD altera a atividade da rede antes ou depois dos neurônios serotoninérgicos, produzindo um resultado dependente de serotonina sem ser, de fato, um fármaco clássico de receptor serotoninérgico.
Essa distinção importa muito. Se um composto acalma o comportamento de um modo bloqueado por um antagonista de 5-HT1A como WAY-100635, isso por si só não prova que o composto é um agonista de 5-HT1A. Prova dependência da sinalização de 5-HT1A naquele modelo. Essas não são a mesma afirmação, e a cobertura sobre cannabinoid frequentemente as mistura.
5-HT1A e a questão da ansiedade
A ligação serotoninérgica mais forte para os cannabinoid, especialmente o CBD, é 5-HT1A. Esse receptor é um receptor de serotonina acoplado a Gi/o, expresso tanto como autorreceptor em neurônios serotoninérgicos da rafe quanto como receptor pós-sináptico em regiões relacionadas à ansiedade, incluindo hipocampo, amígdala e córtex pré-frontal. Fármacos que ativam ou recrutam esse sistema podem reduzir a ansiedade em alguns cenários, mas a localização do receptor importa: reduzir a descarga serotoninérgica via autorreceptores não é a mesma coisa que moldar a sinalização pós-sináptica em circuitos límbicos.
O CBD entrou nessa discussão por meio de trabalhos pré-clínicos dos anos 2000 e 2010 mostrando efeitos semelhantes a ansiolíticos em testes como elevated plus maze, Vogel conflict test e paradigmas de medo contextual, com bloqueio parcial por WAY-100635. Um artigo amplamente citado é Campos e Guimarães, 2008, que encontrou que o CBD intra-prelímbico reduzia respostas cardiovasculares relacionadas ao estresse por restrição e que mecanismos de 5-HT1A contribuíam para o efeito. Outro estudo humano importante é Bergamaschi et al., 2011: em um teste de fala pública simulada, 600 mg de CBD oral reduziram a ansiedade em indivíduos com transtorno de ansiedade social em comparação com placebo. Esse artigo não provou mediação por 5-HT1A em humanos, mas se alinhou ao padrão pré-clínico e ajudou a tornar a serotonina um mecanismo candidato sério, e não uma frase de marketing.
A farmacologia do receptor, porém, nunca se resolveu em uma história simples do tipo “CBD é um agonista de serotonina”. Trabalhos in vitro iniciais sugeriram que o CBD poderia deslocar ligantes em receptores humanos 5-HT1A e se comportar como agonista em alguns ensaios de sinalização, mas as afinidades eram modestas e dependentes do ensaio. Russo e colegas, em 2005, relataram CBD como agonista em receptores humanos clonados de 5-HT1A em ensaios de ligação [35S]GTPγS. Esse achado foi influente, mas trabalhos posteriores complicaram o quadro. Alguns grupos viram atividade direta fraca. Outros viram realce funcional melhor explicado por efeitos alostéricos ou de nível de membrana. A literatura é consistente em um único ponto: 5-HT1A importa mais para a farmacologia do CBD relacionada à ansiedade do que CB1 ou CB2 sozinhos conseguem explicar.
É por isso que o reducionismo de receptor falha. Se o CBD fosse simplesmente um agonista limpo de 5-HT1A, seu perfil se pareceria mais de perto com o de ansiolíticos serotoninérgicos conhecidos do que realmente se parece. Em vez disso, o sinal comportamental é altamente dependente do contexto, muitas vezes mostrando curvas dose-resposta em U invertido. Em alguns testes com roedores, doses moderadas reduzem o comportamento semelhante à ansiedade, enquanto doses mais baixas ou mais altas fazem menos. Isso é um alerta contra histórias de um receptor só. A ativação de TRPV1 em concentrações mais altas é uma razão proposta. O mesmo vale para efeitos sobre o tônus do endocannabinoid, captação de adenosina e manejo intracelular de cálcio. Uma molécula pode recrutar 5-HT1A e ainda assim se recusar a se comportar como um fármaco de 5-HT1A de manual.
Ligação direta versus efeitos serotoninérgicos indiretos
A melhor forma de ler a evidência sobre serotonina é por níveis. No nível molecular, há suporte para interação direta entre CBD e 5-HT1A, mas não do tipo limpo, de alta afinidade e alta eficácia, que encerra a questão. Dependendo do sistema de ensaio, o CBD já foi descrito como agonista fraco, agonista parcial ou modulador alostérico positivo. A discordância não é mera semântica. Agonistas ortostéricos ocupam o sítio principal de ligação da serotonina. Moduladores alostéricos positivos mudam o comportamento do receptor em outro sítio e podem amplificar respostas da serotonina endógena sem ativar fortemente o receptor por conta própria. Esses mecanismos têm implicações diferentes para dose, timing, efeitos colaterais e tradução para humanos.
Os dados de sinalização celular muitas vezes apontam mais para facilitação do que para ativação bruta. Em algumas preparações, o CBD aumenta cascatas de sinalização mediadas por 5-HT1A, incluindo efeitos sobre ERK e outras vias a jusante, mais do que seria previsto pela sua ligação fraca isolada. Há várias explicações possíveis. O CBD é altamente lipofílico e particiona-se em membranas, onde pode alterar o microambiente do receptor e o acoplamento à proteína G. Também pode elevar indiretamente a sinalização de anandamida, e o cruzamento endocannabinoid-serotonina na rafe dorsal e no prosencéfalo é bem documentado. Soma-se a isso a adenosina: o CBD inibe o transporte de nucleosídeos equilibrativo em alguns sistemas, aumentando a adenosina extracelular e alterando a excitabilidade neuronal de maneiras que podem alimentar circuitos serotoninérgicos. Nada disso torna 5-HT1A irrelevante. Torna-o embutido em uma rede.
A farmacologia animal oferece evidências mais fortes de dependência de serotonina do que de agonismo direto. Repetidamente, WAY-100635 atenua os efeitos do CBD em modelos de ansiedade, pânico, náusea e estresse. Resstel et al., 2009, por exemplo, ligaram a atenuação das respostas ao estresse agudo por restrição pelo CBD a mecanismos de 5-HT1A. O trabalho de Rock e Parker sobre náusea e náusea antecipatória em roedores também implicou 5-HT1A no perfil antiemético do CBD. Esses resultados são úteis, mas devem ser lidos como evidência de via. Se bloquear 5-HT1A remove o efeito, a via está envolvida. Isso não resolve se o receptor está sendo ligado diretamente, modulado alostericamente ou recrutado por mudanças em nível de circuito.
A evidência humana continua modesta. O estudo de Bergamaschi de 2011 é frequentemente citado porque mostrou um sinal ansiolítico mensurável em ansiedade social durante fala pública. Estudos de imagem menores relataram que o CBD altera a ativação límbica e paralímbica durante tarefas de processamento emocional. Ainda assim, nenhum desses estudos identificou ocupação do receptor 5-HT1A em humanos da maneira como estudos PET podem fazer para fármacos serotoninérgicos estabelecidos. Essa ausência é importante. Estamos inferindo mecanismo por convergência, não medindo-o diretamente em doses clínicas.
CBD’s calming effects depend in part on 5-HT1A signaling.Limited evidence
Por que o perfil calmante do CBD resiste a rótulos simples de receptor
O CBD já tem um uso aprovado pela FDA, e não é ansiedade. O rótulo de 2024 da FDA para solução oral de cannabidiol limita a indicação a convulsões associadas à síndrome de Lennox-Gastaut, à síndrome de Dravet ou ao complexo de esclerose tuberosa em pacientes com 1 ano ou mais. Esse fato é um bom freio contra exageros. Um composto pode ter sinais ansiolíticos críveis sem que a eficácia em ansiedade esteja resolvida no nível regulatório, e pode ter envolvimento serotoninérgico sem se encaixar de modo limpo na caixa dos fármacos serotoninérgicos.
Parte do problema é a escala. In vitro, cannabinoid são farmacologicamente confusos. In vivo, são ainda mais, porque distribuição, metabolismo, acúmulo tecidual e diferenças entre espécies mudam quais alvos importam. Um efeito de receptor visto a 10 micromolar em células transfectadas pode ser irrelevante após uma dose oral comum, enquanto um efeito aparentemente mais fraco in vitro pode importar se o composto se concentrar em tecido cerebral rico em lipídios ou se metabólitos ativos contribuírem. Essa é uma das razões pelas quais manchetes sobre “o receptor de serotonina que o CBD atinge” costumam correr mais rápido do que os dados.
Outra razão é a biologia de circuitos. Ansiedade não é gerada por um receptor só. Ela emerge de interações entre amígdala, núcleo da estria terminal, córtex pré-frontal medial, hipocampo, hipotálamo e núcleos do tronco encefálico, incluindo a rafe dorsal. O CBD parece deslocar a atividade ao longo dessa rede. Parte desse deslocamento provavelmente recruta 5-HT1A. Parte pode envolver TRPV1, que pode se opor à ansiolise em doses mais altas. Parte pode envolver mudanças no tônus da anandamida mediadas por FAAH, embora a inibição de FAAH pelo CBD em exposições terapêuticas em humanos seja debatida. Parte pode refletir efeitos anti-inflamatórios ou autonômicos que retroalimentam a ansiedade percebida. Uma vez adotada essa visão em rede, a incapacidade de uma explicação com um único rótulo deixa de parecer fraqueza e passa a parecer um relato realista da farmacologia.
É também para isso que o desenvolvimento de fármacos está caminhando. A era da química medicinal está menos interessada em discutir se um composto é “tipo THC” do que em definir combinações de alvos e separar os efeitos desejados da intoxicação. Essa lógica aparece em trabalhos muito além da serotonina, desde o rastreamento estrutural de CB2 no artigo de 2016 do Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, até esforços mais recentes para separar analgesia de prejuízo central. Também aparece em programas ansiolíticos em estágio de empresa. Em 2025, a MIRA Pharmaceuticals disse que seu candidato MIRA-55 mostrava um “differentiated mechanism of action” e “anxiolytic activity relative to THC” em dados pré-clínicos reportados em um comunicado para a Nasdaq. O nível de evidência precisa ficar explícito aqui: pré-clínico, reportado pela empresa, não prova clínica. Ainda assim, o sinal é relevante como indicador de mercado e de pesquisa. Empresas estão ativamente buscando agentes inspirados em cannabinoid que acalmem sem agir como THC, e mecanismos voltados à serotonina fazem parte dessa busca.
O contexto de saúde pública torna isso mais do que uma disputa acadêmica. Em 2025, o HHS afirmou que 7-hydroxymitragynine “poses an imminent hazard to public safety” ao apoiar medidas regulatórias contra produtos perigosos com 7-OH intensificado. Diferentes modificações químicas criam diferentes perfis-alvo e diferentes riscos. A mesma lição vale em todo o espaço dos cannabinoid. Se um produto é tratado como intercambiável com cannabinoid vegetais familiares porque soa próximo de THC ou CBD, a farmacologia é achatada e a avaliação de segurança sofre.
Então, onde as evidências chegam? 5-HT1A é o mecanismo serotoninérgico mais bem sustentado para os efeitos calmantes do CBD, mas a afirmação mais forte que os dados atualmente suportam não é “CBD é um agonista de serotonina”. É mais estreita e mais defensável: o CBD frequentemente produz efeitos semelhantes a ansiolíticos e de amortecimento do estresse que dependem em parte da sinalização de 5-HT1A, enquanto o modo exato de engajamento parece variar conforme o ensaio, a dose, o tecido e o contexto do circuito. Pode ser menos elegante do que um slogan de um receptor só. Também está muito mais perto da verdade.
Além da lista solicitada: canais de sódio e outros alvos não canônicos que já estão mudando a conversa sobre dor
Durante anos, a maior parte das discussões públicas sobre a farmacologia cannabinoid da dor ficou presa em uma história de dois receptores: CB1 explica os efeitos psicoativos, CB2 explica os efeitos imunológicos, e todo o resto é secundário. Esse enquadramento agora é pequeno demais. Mesmo dentro da área mais estreita da dor, os cannabinoid não tocam apenas canais TRP, PPARs, GPCR órfãos ou vias ligadas à serotonina. Eles também interagem com canais de sódio dependentes de voltagem que estão no centro da excitabilidade dos nociceptores. Isso importa porque NaV1.7 e NaV1.8 não são notas laterais periféricas; estão entre as portas moleculares mais estudadas para a sinalização da dor em neurônios sensoriais de pequeno diâmetro.
A mudança é mais do que acadêmica. Desenvolvedores de fármacos gastaram anos tentando bloquear a transmissão da dor no nível dos nervos periféricos sem reproduzir sedação, intoxicação, alteração de memória e potencial de abuso associados à forte ativação central de CB1. Se um cannabinoid, ou um scaffold derivado de cannabinoid, puder atenuar a descarga dos nociceptores ao agir em canais NaV fora do cérebro, isso abre uma lógica terapêutica muito diferente. Leva a conversa de “Quanto ele atinge CB1?” para “Onde ele age, em que concentração e em qual tecido?”
Esse mapa-alvo mais amplo também combina com o momento regulatório maior. Em 2025, o U.S. Department of Health and Human Services avisou que “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety”, lembrando que pequenas mudanças químicas podem produzir farmacologias e perfis de segurança muito diferentes. A política sobre cannabinoid frequentemente ficou atrás desse fato básico. Tratar todos os compostos adjacentes a intoxicantes como se diferenciassem apenas por origem ou por força equivalente a THC perde o ponto. A farmacologia em nível de alvo é o que prevê efeito, risco e potencial de fármaco.
THC inhibits peripheral nociceptors by targeting NaV1.7 and NaV1.8 sodium channels.Preliminary evidence
THC em NaV1.7 e NaV1.8, canais nociceptivos periféricos
A razão mais direta para os canais de sódio agora pertencerem a qualquer mapa sério dos cannabinoid é o relatório de 2025 do grupo da Hebrew University of Jerusalem mostrando que THC inibe nociceptores periféricos ao atingir “NaV1.7 and NaV1.8 nociceptive sodium channels.” Essa é uma expansão significativa do vocabulário do campo. NaV1.7 e NaV1.8 são fortemente expressos em neurônios periféricos de percepção da dor, e seu papel na biologia da dor humana não é especulativo. Mutações de perda de função em NaV1.7 podem produzir insensibilidade congênita à dor; mutações de ganho de função podem causar síndromes dolorosas graves. NaV1.8 está igualmente ligado a estados de dor inflamatória e neuropática porque sustenta disparos repetitivos em nociceptores, especialmente em condições despolarizadas.
Então, quando se mostra que THC inibe esses canais, o achado não pertence à categoria de “efeitos fora do alvo diversos”. Ele aponta para um mecanismo que pode reduzir diretamente a excitabilidade das fibras da dor antes que os sinais cheguem à medula espinhal ou ao cérebro.
Isso é uma classe mecanística diferente das histórias cannabinoid mais conhecidas. TRPV1, reconhecido no trabalho que contribuiu para a parcela do Prêmio Nobel de 2021 de David Julius, pode ser ativado ou dessensibilizado por vários cannabinoid, incluindo CBD e CBG, com efeitos fortemente dependentes da dose e do timing. A sinalização por PPAR-gamma foi invocada para efeitos anti-inflamatórios e metabólicos, muitas vezes com a complicação de que acúmulo intracelular e metabólitos podem importar tanto quanto o composto original. GPR55 continua em debate suficiente para que chamá-lo de “CB3” ainda seja mais slogan do que ciência resolvida. As ligações com serotonina, especialmente 5-HT1A, ajudam a explicar partes do perfil ansiolítico do CBD, mas o circuito é dependente do contexto e frequentemente indireto. A inibição de canais de sódio é menos glamourosa. Também é, para a dor, potencialmente mais prática.
Um ponto-chave aqui é a promiscuidade farmacológica. Os cannabinoid costumam ser ligantes “sujos” no sentido técnico: engajam múltiplos alvos com diferentes afinidades e consequências funcionais. Isso não é uma falha na ciência; é a própria ciência. O THC pode continuar sendo mais conhecido pelo agonismo central de CB1, mas isso não cancela sua capacidade de modular canais iônicos periféricos nas condições adequadas. A pergunta real é se essas condições são alcançáveis in vivo de maneiras que ajudem os pacientes mais do que prejudiquem. O achado da Hebrew University diz que isso é pelo menos plausível o suficiente para merecer atenção séria no desenvolvimento de fármacos.
A cannabinoid analgesic can relieve pain without producing the high through peripheral or non-CB1 mechanisms.Preliminary evidence
Analgesia periférica sem intoxicação central
É aqui que a área da dor fica interessante. Um mecanismo cannabinoid que reduza a descarga dos nociceptores periféricos poderia, em teoria, separar analgesia do comprometimento cognitivo normalmente ligado à ativação de CB1 no cérebro. Essa distinção está no centro do trabalho translacional atual, e não como um benefício lateral.
O relatório de 2026 da ScienceDaily capturou a ideia em linguagem simples: pesquisadores identificaram “a cannabis compound that relieves pain without the high.” Essa formulação deve ser lida com cuidado. É um sinal em estágio de pesquisa, não uma terapia estabelecida, e resumos populares muitas vezes comprimem detalhes mecanísticos. Ainda assim, a importância translacional é óbvia. Se a atividade analgésica puder ser gerada por restrição periférica, baixa penetração cerebral, engajamento seletivo de alvos fora de CB1 ou alguma combinação dos três, então o antigo trade-off entre alívio da dor e intoxicação não é fixo pela natureza. É um problema de química medicinal.
Esse ponto também ajuda a explicar por que o campo passou de rótulos grosseiros de receptor. O artigo de 2016 do ACS Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, reflete uma mudança mais ampla em direção ao desenho estrutural em vez de tratar os cannabinoid como uma família farmacológica com um único eixo útil de variação. Químicos agora perguntam como ajustar forma do scaffold, lipofilicidade, viés de receptor, distribuição tecidual e destino metabólico. O objetivo não é apenas atividade mais forte. É atividade seletiva no lugar certo.
A analgesia periférica é exatamente o tipo de desfecho em que essas distinções importam. Um composto que atravessa mal a barreira hematoencefálica, mas inibe de modo significativo NaV1.7 ou NaV1.8 em nociceptores, pode aliviar dor inflamatória ou neuropática com muito menos intoxicação do que o próprio THC. Isso ainda é uma ambição, não um fato clínico. Mesmo assim, o trabalho da Hebrew University dá a essa ambição um ponto de apoio molecular.[8]Agriculture Improvement Act of 2018. U.S. Congress. Congress.gov, 2018. https://www.congress.gov/bill/115th-congress/house-bill/2/text
Também torna mais nítida a forma como pensamos sobre produtos de cannabis já em circulação. A definição legal de hemp na Farm Bill de 2018 se baseia em teor de delta-9 THC “not more than 0.3 percent on a dry weight basis.” Esse número é regulatório, não farmacológico. Não diz nada sobre canais de sódio, ativação de TRP, sinalização de 5-HT1A, metabólitos ativos ou exposição tecidual. O mesmo vale para intoxicantes mais novos, intensificados ou semissintéticos. Segurança não pode ser inferida a partir da história de origem. Precisa ser inferida a partir de alvos, concentrações e farmacocinética real.
Por que esses achados importam para futuros fármacos cannabinoid
A implicação mais forte da história NaV1.7/NaV1.8 é que futuros medicamentos cannabinoid podem ter sucesso justamente por serem menos “cannabinoid-like” no sentido popular. Ou seja, os descendentes úteis da química da cannabis talvez não sejam fármacos que imitem amplamente o THC fumado. Podem ser compostos que aproveitam parte do scaffold, evitam sinalização central de CB1 e agem, em vez disso, em canais iônicos periféricos ou em conjuntos mistos de alvos não canônicos.
Essa possibilidade já se encaixa no conjunto mais amplo de evidências. O uso aprovado do CBD é para distúrbios convulsivos, não para analgesia rotineira, e mesmo lá sua farmacologia não é bem explicada apenas por CB1 ou CB2. O rótulo da FDA para solução oral de cannabidiol afirma que ela é indicada para convulsões associadas à síndrome de Lennox-Gastaut, à síndrome de Dravet ou ao complexo de esclerose tuberosa em pacientes com 1 ano de idade ou mais. Em outras palavras, o único grande medicamento cannabinoid aprovado pela FDA em uso amplo já resiste à narrativa simplista de receptor. O campo da dor está alcançando a mesma lição.
Programas iniciais de empresas apontam na mesma direção, embora exijam cautela. Em 2025, a MIRA Pharmaceuticals relatou dados pré-clínicos alegando que MIRA-55 mostrou um “differentiated mechanism of action” e “anxiolytic activity relative to THC.” Comunicados de empresa não são evidência neutra, e sinais pré-clínicos muitas vezes fracassam. Mesmo assim, mostram para onde a química medicinal está indo: para longe da imitação desorientada de THC e em direção ao desenho moldado por mecanismo.
Para dor, os canais de sódio podem se tornar um dos ramos mais consequentes dessa estratégia de design. Não o único ramo. TRPV1, TRPA1, PPAR-gamma, GPR55, vias ligadas à adenosina e modulação serotoninérgica continuarão no quadro. Mas NaV1.7 e NaV1.8 trazem algo especialmente atraente: uma conexão direta com o comportamento elétrico das fibras da dor periférica. Isso os torna alvos excepcionalmente concretos em um campo repleto de explicações indiretas.
O resultado é uma forma mais limpa de pensar sobre cannabinoid e dor. Não CB1 versus CB2. Nem planta versus sintético. E não “high” versus “medical” como se fossem categorias moleculares. A distinção melhor é entre mecanismos de intoxicação central e mecanismos úteis perifericamente. O achado da Hebrew University coloca o próprio THC nos dois lados dessa linha. É exatamente por isso que ele importa.
| Cannabinoid | Non-CB1/CB2 targets emphasized in the article | Main caution |
|---|---|---|
| CBD | TRPV1, TRPA1, TRPM8, 5-HT1A, PPAR-gamma, GPR55, adenosine-related signaling | Many signals come from assay-dependent and often micromolar studies |
| THC | TRPV2, reported TRPV1/TRPA1 interactions, NaV1.7, NaV1.8 | Central CB1 effects can dominate and obscure other mechanisms |
| CBG | TRPA1, TRPV1, TRPM8, alpha-2 adrenergic, 5-HT1A-related interactions | Translation from in vitro concentration to human exposure is uncertain |
| CBC | TRPA1, TRPV family channels | Human evidence is thin |
| THCV | Non-CB1 possibilities including TRP and metabolic signaling | Dose and context can change apparent behavior |
| CBDA / THCA | 5-HT1A-related, TRP, enzyme-related actions discussed in preclinical work | Stability, decarboxylation, and exposure complicate interpretation |
Como cannabinoid específicos diferem quando você deixa de perguntar apenas sobre CB1 e CB2
Quando você deixa de tratar CB1 e CB2 como a história inteira, a lista familiar de cannabinoid fica muito menos arrumada. Essas moléculas não são chaves limpas para duas fechaduras. São compostos lipofílicos e sensíveis à concentração que podem atingir canais iônicos, receptores nucleares, processos de transporte, GPCR órfãos e, em alguns casos, canais de sódio dependentes de voltagem. Isso importa porque dor, inflamação, controle de crises, ansiedade e efeitos adversos muitas vezes se encaixam mal em rótulos simples de “agonista de CB1” ou “agonista de CB2”.
O CBD forçou essa mudança mais do que qualquer outro cannabinoid. Sua baixa eficácia clássica em receptores cannabinoid tornou o antigo quadro difícil de defender, especialmente depois que uma solução oral de cannabidiol aprovada pela FDA passou a ter indicações para convulsões associadas à síndrome de Lennox-Gastaut, à síndrome de Dravet e ao complexo de esclerose tuberosa em pacientes com 1 ano de idade ou mais. Um cannabinoid clinicamente útil, com intoxicação limitada do tipo CB1, era um problema para o reducionismo de receptor. Os pesquisadores tiveram de olhar para outros lugares.
O campo mais amplo o seguiu. Trabalhos sobre canais TRP se apoiaram em uma biologia sensorial reconhecida no mais alto nível quando o Prêmio Nobel de Fisiologia ou Medicina de 2021 foi concedido a David Julius e Ardem Patapoutian por descobertas de receptores para temperatura e toque. A farmacologia cannabinoid cruza diretamente essa biologia: TRPV1, TRPA1 e TRPM8 continuam reaparecendo porque muitos phytocannabinoid podem ativá-los, inibi-los ou dessensibilizá-los em concentrações experimentalmente relevantes. Isso não significa que todo resultado de ensaio preveja um efeito humano. Significa que a velha ideia de que a ação “real” dos cannabinoid começa e termina em CB1/CB2 está errada.
CBD: o protótipo da complexidade fora de CB1/CB2
CBD é o melhor exemplo de por que a promiscuidade de alvos importa. Ele tem baixa afinidade e eficácia limitada em CB1 e CB2 em comparação com THC, mas claramente faz algo biologicamente importante. A lacuna entre esses dois fatos gerou uma série de hipóteses de trabalho, algumas mais fortes do que outras.
Canais TRP foram uma das primeiras alternativas sérias. O CBD ativa TRPV1 em sistemas heterólogos, e TRPV1 não é um desvio obscuro; é um canal central na nocicepção e na dor inflamatória. Ativação pode parecer contraintuitiva se o objetivo terapêutico for analgesia, mas a ativação repetida ou sustentada de TRPV1 frequentemente produz dessensibilização, reduzindo a excitabilidade subsequente. Essa é uma razão pela qual a farmacologia TRP pode parecer contraditória no papel. Um composto pode ativar primeiro e silenciar o sistema depois. O CBD também mostra ações em TRPA1 e pode inibir TRPM8 em alguns modelos, tornando-o farmacologicamente mais amplo do que o resumo público usual de “cannabinoid não intoxicante”.
A história da serotonina é mais contestada, mas ainda importante. Uma literatura pré-clínica extensa liga o CBD a efeitos relacionados a 5-HT1A, especialmente em modelos de ansiedade, estresse e náusea. A afirmação mais limpa não é que o CBD seja um agonista simples de alta afinidade para 5-HT1A do modo como se discute a farmacologia do buspirona, mas que a sinalização de 5-HT1A frequentemente contribui para os efeitos do CBD in vivo, às vezes por agonismo parcial, às vezes por mecanismos alostéricos ou em nível de circuito que ainda não estão resolvidos. Essa distinção importa. Muitos resumos achatam “envolve 5-HT1A” em “funciona por serotonina”. Os dados não sustentam essa simplificação.
PPAR-gamma é outro candidato recorrente, e aqui a química intracelular importa. PPARs são receptores nucleares, então uma molécula lipofílica que particiona em membranas e células pode afetá-los de formas que um modelo centrado em receptores de superfície não capta. O CBD já foi relatado como ativador de PPAR-gamma em sistemas celulares, e a sinalização de PPAR-gamma tem ligações plausíveis com inflamação, metabolismo lipídico, fibrose e neuroinflamação. Mas há um porém: alguns efeitos relacionados a PPAR podem refletir metabólitos, exposição mais longa ou mudanças indiretas em mediadores lipídicos endógenos, em vez de ocupação direta do receptor em um passo único. A farmacologia é real o suficiente para ser estudada seriamente, mas mais fraca como slogan do que como mecanismo.
A sinalização da adenosina entrou na conversa porque o CBD parece inibir o transporte de nucleosídeos em alguns sistemas, potencialmente elevando o tônus extracelular de adenosina e afetando indiretamente vias anti-inflamatórias ligadas a A2A. Novamente, isso não é farmacologia de ligação a receptor organizada e limpa. É farmacologia de transporte e de contexto tecidual. Se isso soa mais bagunçado do que “CBD atinge CB2”, é porque é. Também é mais plausível.
Então há GPR55, muitas vezes apresentado como um suposto “CB3.” Esse rótulo ainda é confiante demais. O CBD pode antagonizar ou modular de outras formas a sinalização ligada a GPR55 em vários sistemas experimentais, e GPR55 foi implicado em excitabilidade, biologia óssea, inflamação e vias relacionadas ao câncer. Mas os ligantes endógenos do receptor, o acoplamento de via e a relevância translacional continuam debatidos. GPR55 é um gerador útil de hipóteses, não um substituto resolvido para CB1/CB2.
O resultado é simples: o CBD tornou-se o protótipo da farmacologia cannabinoid fora de CB1/CB2 porque seus efeitos clinicamente relevantes não podiam ser explicados de outra forma. Isso continua verdadeiro.
CBG, CBC, THCV, cannabinoid ácidos e a farmacologia dos minor cannabinoids
Os minor cannabinoids costumam ser vendidos ao público como se cada um tivesse uma única característica de personalidade. A farmacologia não coopera.
O CBG costuma ser descrito como levemente ativo em CB1 e CB2, mas os sinais mais interessantes estão fora desses receptores. Ele interage com sistemas alpha-2 adrenérgicos e 5-HT1A em alguns ensaios, mostra atividade em canais TRP e já foi estudado por efeitos anti-inflamatórios e analgésicos que não se reduzem de forma limpa à ativação clássica de receptores cannabinoid. Ele também ilustra um problema recorrente: atividade in vitro em faixa micromolar é fácil de publicar e difícil de traduzir. Um acerto em receptor a 10 micromolar pode importar numa placa, mas não no plasma humano, ou só em tecidos onde o composto se concentra.
CBC é há muito pouco caracterizado em relação ao entusiasmo que o cerca. Parece envolver canais da família TRPV e TRPA1 de forma mais convincente do que CB1, e alguns estudos sugerem efeitos anti-inflamatórios ou semelhantes a analgésicos em animais. Também há interesse na interação do CBC com o tônus endocannabinoid, incluindo efeitos que poderiam alterar indiretamente a sinalização da anandamida. Mas “CBC funciona por canais TRP” ainda é um ponto de partida, não uma resposta final. As evidências humanas são muito frágeis.
THCV é um caso mais complicado porque pode se comportar de maneira diferente por dose e contexto até mesmo em CB1, sendo frequentemente descrito como antagonista neutro ou ligante de baixa eficácia em algumas concentrações e como um composto mais agonista em outras. Fora de CB1/CB2, o THCV foi relacionado a ações em TRP e a efeitos metabólicos, com interesse repetido em apetite, controle glicêmico e balanço energético. Parte desse entusiasmo ultrapassou as evidências anos atrás. A melhor interpretação é que o THCV é farmacologicamente interessante justamente porque não se encaixa no modelo de THC, não porque algum alvo secundário já tenha explicado completamente seu perfil.
Cannabinoid ácidos merecem mais atenção do que recebem. THCA e CBDA são frequentemente tratados como apenas precursores “crus”, mas sua farmacologia não é simplesmente uma espera inativa até a descarboxilação. O CBDA tem algumas das evidências mais intrigantes para efeitos anti-náusea relacionados a 5-HT1A em trabalhos pré-clínicos, e ambos os cannabinoid ácidos mostraram ações em TRP e relacionadas a enzimas in vitro. Sua menor penetração cerebral em comparação com cannabinoid neutros pode até ser uma vantagem para certos alvos periféricos ou gastrointestinais. O problema é a qualidade dos dados e o realismo de dose. Muitas alegações dependem de estudos animais esparsos ou ensaios celulares com relevância humana incerta.
É aqui que a política atual e a química dos produtos colidem com a farmacologia. A Farm Bill de 2018 definiu hemp usando um limiar de delta-9 THC de não mais que 0,3% em base de peso seco. Essa fronteira legal não diz nada sobre GPR55, TRPV1, NaV1.7, metabólitos ou análogos semissintéticos. Os reguladores começaram a reagir a esse desalinhamento em áreas adjacentes. Em 2025, o HHS disse que “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” ao apoiar a ação da DEA sobre produtos com 7-OH intensificado. Classe de droga diferente, mesma lição: produtos modificados ou enriquecidos podem ter perfis de segurança que divergem bruscamente do material de origem. Os cannabinoid não estão isentos dessa lógica.
Metabólitos, alegações de entourage e o problema da química
Uma razão pela qual a farmacologia cannabinoid fica escorregadia é que o composto original nem sempre representa toda a exposição. Metabólitos podem importar, às vezes muito. O público aprendeu isso principalmente por meio dos metabólitos intoxicantes do THC, mas o princípio mais amplo se aplica à ciência cannabinoid como um todo. Via de administração, metabolismo de primeira passagem, partição tecidual e diferenças entre espécies podem alterar quais alvos são realmente engajados.
Até mesmo o THC, o composto mais fortemente associado a CB1, não se restringe à biologia de CB1. Em 2025, pesquisadores da Hebrew University relataram que o THC inibe nociceptores periféricos ao atingir canais de sódio nociceptivos NaV1.7 e NaV1.8. Isso lembra fortemente que a analgesia pode envolver efeitos diretos sobre a maquinaria de excitabilidade, e não apenas sinalização por GPCR. Um relatório de pesquisa de 2026 destacado pela ScienceDaily levou a mesma ideia translacional mais adiante, descrevendo um composto derivado de cannabis que aliviou a dor sem o high. Isso continua sendo evidência em estágio de pesquisa, não medicina estabelecida, mas aponta para uma estratégia séria de desenvolvimento de fármacos: separar analgesia da intoxicação central explorando alvos fora de CB1, restrição periférica ou ambos.
A química medicinal está se movendo nessa direção. O rastreamento estrutural em torno de receptores cannabinoid já estava bem estabelecido pelo artigo de 2016 do Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, e esforços mais recentes buscam cada vez mais mecanismos diferenciados, em vez de compostos genéricos “tipo THC, mas mais fracos”. Um comunicado de 2025 da MIRA Pharmaceuticals, que deve ser tratado como dados pré-clínicos reportados pela empresa e não como confirmação independente, descreveu o MIRA-55 como tendo um mecanismo diferenciado e atividade ansiolítica relativa ao THC. O ponto importante não é que a alegação esteja provada. É que desenvolvedores de fármacos agora presumem que a separação de alvos importa.
Isso nos leva ao entourage effect. Como ideia científica estreita, é plausível que combinações de cannabinoid, terpenes e metabólitos possam alterar farmacocinética ou produzir efeitos aditivos, opostos ou, ocasionalmente, supra-aditivos em múltiplos alvos. Como explicação universal para por que qualquer produto misto de cannabis supostamente funciona melhor, é geralmente vaga demais para testar e elástica demais para falsificar.
O problema da química é básico. Uma mistura contendo THC, CBD, CBG, cannabinoid ácidos, produtos oxidados, terpenes residuais e metabólitos variáveis não é uma única intervenção. É um conjunto de peças móveis cujo efeito depende de proporções de concentração e do tempo. Existe farmacologia plausível multi-alvo. Existe também simplificação não sustentada. Essas coisas não são iguais.
O padrão melhor é evidência específica ao alvo vinculada à exposição. Qual composto, em que concentração, em qual tecido, produzindo que efeito mensurável? Quando você faz essas perguntas, a mitologia se dissipa e a verdadeira história dos cannabinoid entra em foco: não dois receptores, mas um mapa farmacológico lotado.
Os métodos importam: por que o desenho do ensaio molda o que pensamos que os cannabinoid fazem
As alegações sobre alvos cannabinoid costumam soar mais limpas do que os dados que as sustentam. Um artigo diz que o CBD “ativa TRPV1”, outro diz que ele é um “agonista de 5-HT1A”, um terceiro chama GPR55 de candidato a receptor cannabinoid, e um estudo de docking propõe uma pose organizada em CB2 ou em algum bolso de GPCR órfão. Essas declarações podem ser úteis em termos de direção. Não são todas o mesmo tipo de evidência.
Essa distinção importa porque cannabinoid são moléculas gordurosas, amantes de membrana, com o hábito ruim de parecer ativas em mais lugares do que realmente importam in vivo. O campo aprendeu isso da maneira difícil. Se um composto se particiona em membranas, altera propriedades da bicamada, se acumula dentro das células, forma metabólitos ativos ou só mostra efeito em 10 a 50 micromolar, pode gerar histórias de alvo que desmoronam quando testadas sob condições mais rigorosas. Para a farmacologia da cannabis, os métodos não são um detalhe técnico. Eles decidem quais mecanismos sobrevivem.
Ensaios de ligação, ensaios funcionais e estudos de docking
Comece com a pergunta mais antiga e limpa: o composto se liga? Em um ensaio de ligação com radioligante, membranas ou células intactas que expressam um receptor são incubadas com um ligante radioativo conhecido mais concentrações crescentes do composto de teste. Se a molécula de teste desloca o radioligante, os investigadores estimam a afinidade, muitas vezes relatada como Ki. Isso é útil. Também é limitado. Ligação diz que uma molécula pode ocupar um sítio sob condições de ensaio; não diz o que acontece depois.
É por isso que os ensaios funcionais importam mais do que a ligação quando as pessoas fazem alegações sobre sinalização. Para canais TRP como TRPV1, TRPA1 ou TRPM8, os pesquisadores costumam usar ensaios de fluxo de cálcio com corantes fluorescentes. Se a abertura do canal deixa o cálcio entrar na célula, a fluorescência aumenta. O apelo é óbvio: esses ensaios escalam bem e podem comparar muitos compostos rapidamente. O problema é igualmente óbvio se você conhece cannabinoid. Alguns cannabinoid são fluorescentes, alguns perturbam membranas, alguns liberam cálcio indiretamente de estoques intracelulares e alguns desencadeiam dessensibilização do canal após ativação inicial. Um único pico em uma curva de cálcio pode esconder vários mecanismos.
Patch clamp é mais lento, mas muito mais informativo. Ele registra corrente iônica diretamente. Para canais iônicos, especialmente canais de sódio como NaV1.7 e NaV1.8, patch clamp pode mostrar se o fármaco altera ativação, inativação, probabilidade de abertura ou densidade de corrente. É por isso que o relatório de 2025 da Hebrew University sobre THC atuando em nociceptores periféricos via NaV1.7 e NaV1.8 é metodologicamente importante: eletrofisiologia direta pode separar um efeito real em canal de um sinal celular vago. Se um cannabinoid reduz corrente de sódio em nociceptores em concentrações atingidas no tecido periférico, isso tem mais peso mecanístico para analgesia do que outro rótulo frouxo de “interação com receptor”.
GPCRs e receptores nucleares exigem leituras diferentes. Para GPCRs, os investigadores podem medir cAMP, recrutamento de beta-arrestina, ligação de GTPγS, fosforilação de ERK ou sinais de cálcio em linhagens celulares engenheiradas. Esses métodos não são intercambiáveis. Um cannabinoid pode parecer agonista em uma via, agonista parcial em outra e antagonista em uma terceira. Isso não é descuido; é viés de sinalização. Para PPAR-gamma, a abordagem comum é um ensaio repórter em que um promotor responsivo a PPAR dirige luciferase. A luz aumenta, e o composto é chamado de ativador de PPAR. Mas ensaios repórter estão vários passos afastados do engajamento direto do alvo. Um cannabinoid lipofílico pode alterar a transcrição indiretamente por estresse celular, metabolismo ou mudanças em mediadores lipídicos endógenos.
Leituras de expressão gênica estão ainda mais a jusante. Se um artigo mostra que o CBD altera transcritos inflamatórios e o efeito é reduzido por um antagonista de PPAR, isso é sugestivo, não definitivo. O antagonista pode ser imperfeito. As células podem produzir metabólitos. O cannabinoid pode estar alterando o tônus da adenosina, o manejo de cálcio, o estado redox ou a ordem de membrana. Mecanismo por subtração é frágil.
Depois vem o docking. Docking pergunta se uma pequena molécula pode se encaixar em um bolso de ligação modelado ou determinado experimentalmente e obter uma pontuação favorável. Quando bem usado, é triagem de química medicinal. Quando mal usado, vira certeza decorativa. O artigo de 2016 do ACS Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, é um bom ponto de entrada porque mostra a lógica no seu melhor: triagem orientada por estrutura pode ajudar a priorizar scaffolds, identificar contatos plausíveis receptor-ligante e orientar síntese em torno da seletividade para CB2. Para isso o docking serve. Ele gera hipóteses. Não prova que um phytocannabinoid engaja um alvo em tecido vivo, muito menos que a interação explica analgesia, ansiedade ou inflamação.
Diferenças entre espécies, metabólitos e efeitos de membrana
Mesmo sinais fortes in vitro podem falhar fora da placa de ensaio porque a farmacologia cannabinoid depende fortemente do contexto. Receptores humanos e de roedores nem sempre são funcionalmente idênticos. Um composto ativo em readouts de mouse TRPA1 ou de ratos ligados a 5-HT1A pode mudar de potência ou eficácia no ortólogo humano. O mesmo alerta vale para variantes de splicing, reserva de receptor e background celular. Uma linhagem de receptor superexpressa pode fazer um ligante fraco parecer importante.
O metabolismo acrescenta outra camada. Muitos cannabinoid não permanecem muito tempo na forma original. THC vira 11-hydroxy-THC; outras estruturas formam metabólitos oxidados ou conjugados que podem diferir muito no perfil de alvo. Reguladores estão prestando mais atenção a esse problema em debates adjacentes sobre política de drogas. Em 2025, o HHS afirmou que “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety”, destacando uma lição mais ampla: intoxicantes intensificados ou metabolicamente favorecidos podem se comportar de forma muito diferente do composto original. O espaço da cannabis tem problemas paralelos com cannabinoid semissintéticos, novos isômeros e formulações projetadas para alterar a exposição. Se você ensaia apenas a molécula original, pode perder a espécie que de fato está dirigindo os efeitos in vivo.
Membranas são o maior confusor silencioso. Cannabinoid são lipofílicos o suficiente para se acumularem em bicamadas e compartimentos intracelulares. Isso significa que a concentração nominal no banho costuma ser um mau proxy para a concentração no alvo. Uma aplicação de 10 micromolar em um ensaio celular pode criar uma carga local de membrana muito alta, que pode alterar gating de canal ou comportamento de receptor de forma inespecífica. Também pode produzir falsos negativos se a concentração livre em água for muito menor do que o esperado porque o composto adere ao plástico, a proteínas séricas ou à própria membrana.
Essa é uma das razões pelas quais alegações de alta concentração merecem ceticismo. Se um cannabinoid só afeta um alvo acima de 20 ou 30 micromolar, a primeira pergunta deve ser se isso reflete uma interação fisiologicamente relevante ou um artefato mediado por membrana. Os canais TRP são especialmente vulneráveis a exagero aqui. São alvos genuinamente responsivos a cannabinoid em alguns casos, mas os efeitos podem ser bifásicos, dessensibilizantes rapidamente e altamente sensíveis à concentração. Um breve pico de cálcio em alta faixa micromolar não significa automaticamente um mecanismo terapeuticamente relevante.
De artigos de docking do ACS à farmacologia real
A química medicinal prospera com simplificação, mas a biologia pune a simplificação excessiva. O artigo de docking de CB2 do ACS ilustra a versão útil da simplificação: começar com uma estrutura de receptor, rastrear bibliotecas, priorizar candidatos, sintetizar análogos, testá-los em ensaios de ligação e funcionais e aprender com as relações estrutura-atividade. Essa sequência pode gerar fármacos reais. Também pode revelar que a pose in silico mais atraente pertence a um composto com baixa permeabilidade, metabolismo instável, sinalização enviesada ou nenhuma atividade relevante em células nativas.
A distância entre a pose docked e a farmacologia é exatamente onde muitas alegações sobre cannabinoid falham. Uma imagem de docking de CBD ou CBG em TRPV1, 5-HT1A, GPR55 ou PPAR-gamma não é evidência de que o composto dirige a fisiologia relevante em animais ou humanos. Farmacologia real precisa de convergência. Idealmente, isso significa engajamento direto do alvo em concentrações plausíveis, efeitos funcionais em sistemas nativos, evidência de perda de função usando antagonistas ou knock-outs, suporte farmacocinético e algum vínculo com comportamento ou desfecho clínico.
Quando essas camadas se alinham, a história fica muito mais forte. A atual busca translacional para separar analgesia de intoxicação depende dessa lógica. O relatório da ScienceDaily de 2026 descrevendo “a cannabis compound that relieves pain without the high” é interessante precisamente porque aponta além da equivalência grosseira com THC e em direção a mecanismos seletivos por alvo ou restritos perifericamente. O mesmo vale para programas pré-clínicos como o MIRA-55, que um comunicado de 2025 da empresa descreveu como tendo um “differentiated mechanism of action” com atividade ansiolítica relativa ao THC. Declarações de empresa estão no nível baixo da escada de evidências. Ainda assim, refletem a direção do campo: afastar-se de rótulos simples CB1/CB2 e ir para polifarmacologia desenhada, que pode ser medida, e não assumida.
Os leitores devem exigir essa mesma disciplina de artigos acadêmicos sobre cannabinoid. Pergunte qual ensaio foi usado. Pergunte se a concentração ativa é realista. Pergunte se o efeito sobrevive à eletrofisiologia, aos antagonistas, aos knock-outs, aos estudos de metabolismo e à tradução entre espécies. Acima de tudo, pergunte se a alegação de alvo explica o organismo, e não apenas a placa. É assim que a farmacologia cannabinoid fora de CB1/CB2 deixa de ser um conjunto de pistas interessantes e se torna mecanismo real.
Níveis de evidência: da placa de células à clínica
A literatura sobre alvos fora de CB1/CB2 é rica porque os cannabinoid são quimicamente promíscuos. Uma única molécula pode tocar TRPV1, sinalização ligada a 5-HT1A, PPAR-gamma, GPR55, o tônus da adenosina e canais de sódio dependentes de voltagem, dependendo da concentração, do tecido e do perfil de metabólitos. Isso rende artigos mecanísticos interessantes. Não produz, automaticamente, medicina comprovada. Se existe uma regra que mantém esse campo honesto, é simples: cada degrau acima na escada de evidências elimina uma grande parcela de alegações que pareciam convincentes lá embaixo.
O que as evidências pré-clínicas podem e não podem provar
Na base da escada estão ensaios de ligação, registros de canais, sistemas repórter e culturas celulares. Esses métodos são indispensáveis. Foi assim que os pesquisadores aprenderam que a farmacologia cannabinoid vai muito além dos receptores clássicos, e é por isso que explicações de cannabis centradas apenas em receptores hoje parecem datadas. O Prêmio Nobel de 2021 reconheceu David Julius e Ardem Patapoutian “for their discoveries of receptors for temperature and touch”, lembrando que a biologia TRP não é trivia periférica; ela fica perto do centro da ciência moderna da dor. Quando cannabinoid ativam ou dessensibilizam TRPV1, TRPA1 ou canais relacionados em uma placa, isso importa.
Mas uma placa não pode dizer se o mesmo engajamento de alvo ocorre em doses humanas toleradas. Muitos efeitos in vitro de cannabinoid aparecem apenas em concentrações micromolares. Os níveis plasmáticos após dose real podem ser mais baixos, mais transitórios ou deslocados para metabólitos com farmacologia diferente. CBD é o exemplo clássico. Ele mostra repetidamente ações difíceis de explicar apenas por CB1 ou CB2, razão pela qual TRPV1, 5-HT1A, PPAR-gamma, GPR55 e vias da adenosina continuam aparecendo na literatura. Ainda assim, o fato de o CBD modular um alvo em células cultivadas não prova que esse mecanismo dirige um desfecho clínico em epilepsia, ansiedade, dor ou inflamação.
Modelos animais estão um passo mais perto da medicina, mas ainda longe dela. Estudos em roedores podem mostrar que um cannabinoid reduz alodinia, suprime marcadores inflamatórios ou altera comportamento semelhante à ansiedade. Eles podem até sustentar histórias de alvo específico com antagonistas, knock-outs ou restrição periférica. O recente relatório da Hebrew University é um bom exemplo de por que esse trabalho é empolgante: os pesquisadores relataram que THC inibe nociceptores periféricos ao atingir canais de sódio nociceptivos NaV1.7 e NaV1.8. Esse achado contraria a suposição preguiçosa de que a analgesia relacionada ao THC precisa ser uma história de CB1 mais intoxicação. Se o efeito se mantiver, ele sugere ação relevante para dor no próprio nível da excitabilidade periférica.
Ainda assim, mesmo um bom trabalho em animais não pode provar que bloquear NaV1.7/NaV1.8 com um cannabinoid vai se tornar um analgésico humano seguro e eficaz. Diferenças entre espécies importam. Dose importa. Via importa. Leitores comportamentais em camundongos podem enganar. Um sinal de dor amortecido em uma preparação de nervo ou em um ensaio de formalina pode não se traduzir em alívio para neuropatia, osteoartrite ou dor pós-cirúrgica em pessoas. A mesma cautela se aplica a anúncios de empresas. Em 2025, a MIRA Pharmaceuticals disse que seu candidato MIRA-55 mostrou um “differentiated mechanism of action” e “anxiolytic activity relative to THC” em dados pré-clínicos. Isso é legítimo como evidência em estágio de pesquisa. Não é uma alegação de tratamento, e não deve ser tratada como tal.
A mesma cautela translacional vale para manchetes atraentes. Em 2026, a ScienceDaily destacou trabalho sobre “a cannabis compound that relieves pain without the high.” Talvez. Essa é uma direção útil para o desenho de fármacos, especialmente se alvos periféricos ou mecanismos fora de CB1 puderem separar analgesia de intoxicação central. Mas “sem o high” em um relatório pré-clínico é uma hipótese em teste, não um fato estabelecido sobre terapia humana.
Medicamento cannabinoid aprovado e a estreiteza das indicações atuais
Agora vá para o topo da escada: evidência de medicamento aprovado. Aqui o campo fica muito mais estreito. O exemplo mais claro nos EUA é a solução oral de cannabidiol. O rótulo aprovado pela FDA afirma que ela é indicada “for the treatment of seizures associated with Lennox-Gastaut syndrome, Dravet syndrome, or tuberous sclerosis complex in patients 1 year of age and older.” Essa frase é mais informativa do que dezenas de alegações vagas de bem-estar porque nomeia a forma farmacêutica, o desfecho, as doenças e a faixa etária.
Observe o que o rótulo não diz. Não diz que o CBD é aprovado para dor, ansiedade generalizada, insônia, doença inflamatória intestinal, neuroproteção ou “equilíbrio do endocannabinoid” em sentido amplo. Não valida todo mecanismo proposto na literatura pré-clínica. Aprovação nos diz que um produto específico de cannabidiol demonstrou eficácia e segurança aceitável para distúrbios convulsivos específicos em estudos controlados. Não resolve se TRPV1, GPR55, 5-HT1A, PPAR-gamma, efeitos intracelulares de cálcio ou ações de metabólitos são o mecanismo clínico dominante. O mecanismo pode continuar parcialmente sem resolução mesmo quando a eficácia é real.
Essa lacuna entre indicação aprovada e folclore mecanístico é comum em farmacologia. Muitos fármacos funcionaram em pacientes antes que seu mapa completo de alvos fosse entendido. Os cannabinoid não são estranhos nesse ponto. O que é estranho é a frequência com que alegações amplas são reconstruídas a partir de achados dispersos de receptores e então faladas como se já tivessem passado por teste clínico.
A estreiteza das indicações atuais também importa para discussões de saúde pública. Os reguladores distinguem cada vez mais cannabinoid vegetais familiares de intoxicantes alterados, intensificados ou semissintéticos com perfis de risco diferentes. Em 2025, o HHS afirmou que “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” ao apoiar medidas regulatórias sobre produtos com 7-OH intensificado. A declaração dizia respeito a produtos relacionados ao kratom, e não à cannabis, mas a lição regulatória é perfeitamente transferível: quando fabricantes começam a enriquecer, modificar ou sintetizar moléculas psicoativas potentes, o atalho baseado na origem vegetal se torna pouco confiável. A farmacologia em nível de alvo passa a importar muito.
Por que as histórias de mecanismo muitas vezes correm à frente dos dados clínicos
Elas correm à frente porque histórias mecanísticas são rápidas, vívidas e publicáveis. A prova clínica é lenta e cara. Um artigo celular pode mostrar que um cannabinoid ativa TRPA1, antagoniza GPR55 ou muda a sinalização de 5-HT1A em um projeto de duração relativamente curta. Um ensaio randomizado convincente para dor crônica pode levar anos e ainda falhar porque o tamanho do efeito é pequeno, os efeitos adversos limitam a dose ou o alvo pré-clínico nunca foi de fato engajado em pacientes.
A química cannabinoid também convida à interpretação excessiva. Compostos estruturalmente relacionados podem ter perfis-alvo muito diferentes, e o metabolismo pode refazer o quadro após a administração. A via muda a história de novo. Dose oral produz metabólitos de primeira passagem; a inalação altera a cinética; formulações tópicas ou periféricas podem favorecer alvos locais sobre alvos centrais. Até a categoria legal “hemp” diz pouco em termos farmacológicos. A Farm Bill de 2018 traçou a linha em delta-9 THC de “not more than 0.3 percent on a dry weight basis”, um limite estatutário, não biológico.
Portanto, a leitura correta da evidência fora de CB1/CB2 não é nem rejeição nem entusiasmo exagerado. A literatura pré-clínica mostra, de fato, que os cannabinoid agem em mais do que CB1 e CB2. Para dor especialmente, canais TRP e canais de sódio merecem atenção séria. Para CBD, alvos não clássicos não são notas laterais opcionais; provavelmente são centrais para explicar seu perfil. Mas plausibilidade não é eficácia, engajamento de alvo não é benefício para o paciente, e a aprovação de um medicamento cannabinoid para um pequeno conjunto de distúrbios convulsivos não ratifica a nuvem muito maior de alegações construída a partir de diagramas de receptor, comportamento em camundongos e resultados de placa.
Segurança, regulação e por que a farmacologia fora do alvo importa em saúde pública
Problemas de saúde pública não se alinham de forma limpa com diagramas de receptor. Um cannabinoid pode ser derivado de planta, de hemp, semissintético ou totalmente sintético e ainda assim acabar produzindo riscos mal previstos por seu rótulo, sua origem ou sua categoria legal. Esse é o significado prático da farmacologia fora do alvo. Uma vez que uma molécula toca canais TRP, receptores de serotonina, PPARs, sistemas de sinalização semelhantes a GPR55, canais de sódio e outros alvos fora de CB1/CB2, o perfil de segurança pode mudar de maneiras relevantes para vigilância de envenenamento, padrões de produto, políticas sobre direção sob efeito e risco de dependência.
O erro não é apenas científico. É regulatório. A política sobre cannabis muitas vezes tratou “cannabinoid” como se isso já fosse uma explicação.
Intoxicantes intensificados e a lição de política do 7-OH
O alerta atual mais claro nem vem de um constituinte clássico da cannabis. Em 2025, o U.S. Department of Health and Human Services afirmou que “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” ao apoiar a ação da DEA sobre produtos com 7-OH intensificado. Essa frase importa porque mostra autoridades federais de saúde traçando uma linha entre exposição botânica familiar e intoxicantes concentrados ou quimicamente manipulados que podem se comportar de modo muito diferente em populações reais.
A lição política se transfere diretamente para os cannabinoid. Um termo amplo como “derivado de hemp”, “de origem vegetal” ou até “cannabinoid-like” diz quase nada aos reguladores sobre quais alvos um composto engaja em concentrações relevantes para uso. Também diz quase nada aos consumidores sobre potência, início de ação, duração, risco de interação ou potencial de abuso.
Produtos com 7-OH intensificado se tornaram um problema de saúde pública porque a química alterou a exposição. Quando um mercado vai da ocorrência natural em traços ao ingrediente ativo concentrado, a farmacologia deixa de ser uma curiosidade e vira a questão central de segurança. O mesmo padrão já apareceu em torno de cannabinoid semissintéticos e estruturalmente alterados vendidos sob o guarda-chuva da legalidade do hemp após a Farm Bill de 2018 definir hemp por concentração de delta-9 THC de “not more than 0.3 percent on a dry weight basis.” Esse limite por peso seco é uma definição agrícola, não um padrão de farmacologia. Ele não avalia atividade em TRP, bloqueio de canais de sódio, sinalização de 5-HT1A, efeitos de GPR55 ou metabólitos com maior persistência e diferente penetração no sistema nervoso central.
É por isso que efeitos fora do alvo não são um rodapé obscuro. São uma das vias pelas quais produtos comercializados como próximos da cannabis podem gerar danos inesperados. Um composto apresentado ao público como “THC-like but legal” pode diferir em eficácia intrínseca em CB1, mas também pode diferir de maneiras muito menos visíveis: maior estimulação cardiovascular, maior potencial pró-convulsivante ou ansiogênico, disforia mais intensa, sedação incomum ou um perfil tóxico moldado pelo metabolismo e não apenas pelo fármaco original. Essas possibilidades não são hipotéticas em abstrato; são exatamente por que autoridades de saúde reagem de forma diferente a intoxicantes intensificados do que a grandes categorias botânicas.
Uma resposta política sólida começa com perguntas em nível de alvo. Quais receptores e canais são engajados? Em que concentrações? Em quais tecidos? Quais são os principais metabólitos? Há evidência de inibição de canais de sódio que possa alterar nocicepção ou condução cardíaca? A ativação de TRPV1 provavelmente dessensibiliza a sinalização de dor ou, em baixas exposições ou exposições transitórias, irrita e piora sintomas? Essas são perguntas mais difíceis do que “isso é um cannabinoid”, mas são as corretas.
Os limites do pensamento em equivalência com THC
A equivalência com THC é atraente porque simplifica lei, tributação, rotulagem e discussões sobre comprometimento. Também costuma estar errada. Dois compostos podem produzir algum grau de intoxicação e ainda assim diferir fortemente em risco de ansiedade, potencial psicotomimético, efeito analgésico, resposta da frequência cardíaca, controle de emese, desenvolvimento de tolerância e carga de abstinência porque não compartilham o mesmo mapa-alvo mais amplo.
Até mesmo o THC não é farmacologicamente exaurido por CB1 e CB2. Em 2025, pesquisadores da Hebrew University relataram que o THC inibe nociceptores periféricos ao atingir canais de sódio nociceptivos NaV1.7 e NaV1.8. Esse achado vai contra a narrativa simplista “efeito de THC=efeito de CB1”. Se até mesmo um cannabinoid psicoativo canônico pode afetar diretamente canais de sódio dependentes de voltagem relevantes para a dor, então segurança e eficácia não podem ser inferidas apenas pela marca cannabis. Dose, via, distribuição e exposição tecidual tornam-se decisivas.
O mesmo ponto aparece na direção oposta com o CBD. Sua solução oral aprovada é indicada pela FDA para convulsões associadas à síndrome de Lennox-Gastaut, à síndrome de Dravet ou ao complexo de esclerose tuberosa em pacientes com 1 ano ou mais, um fato clínico que nunca foi bem explicado por agonismo de CB1 porque o CBD não é um intoxicante CB1 simples. Seu perfil há muito empurra os pesquisadores para outros mecanismos, incluindo TRPV1, 5-HT1A, sinalização relacionada à adenosina e PPAR-gamma. Nem todos esses mecanismos estão igualmente estabelecidos em humanos, mas juntos tornam inevitável um ponto: os efeitos dos cannabinoid muitas vezes ficam em uma rede, não em um único interruptor de receptor.
Essa perspectiva em rede também explica por que a intoxicação é uma métrica ruim para segurança pública. Um produto pode ser menos intoxicante do que THC e, ainda assim, produzir interações medicamentosas preocupantes, efeitos sobre enzimas hepáticas, estresse cardiovascular, reações de pânico ou sedação. Também pode ser mais intoxicante sem ser mais previsível. O público muitas vezes ouve “mais fraco que THC” ou “mais forte que THC” como se isso encerrasse a questão. Não encerra. Essas expressões geralmente descrevem um fenótipo saliente, não um perfil toxicológico completo.
O pipeline de pesquisa já está avançando além da equivalência com THC. Um comunicado da Nasdaq de 2025 da MIRA Pharmaceuticals descreveu dados pré-clínicos do MIRA-55 alegando um “differentiated mechanism of action” e atividade ansiolítica relativa ao THC. Comunicados de empresa são evidência fraca em comparação com dados clínicos revisados por pares, então a alegação deve ser tratada com cautela. Ainda assim, a direção é real: a química medicinal está tentando separar os efeitos desejados da intoxicação central por meio da alteração do engajamento de alvo. A mesma lógica translacional aparece em um relatório da ScienceDaily de 2026 descrevendo um trabalho sobre “a cannabis compound that relieves pain without the high.” Achados em estágio de pesquisa não são prova clínica, mas reforçam um ponto regulatório central. Se efeitos benéficos podem ser separados da intoxicação, então os danos também podem ser separados da intoxicação. Um produto de “baixo high” não é automaticamente um produto de baixo risco.
Por que cannabinoid novos exigem escrutínio em nível de alvo
Cannabinoid novos merecem mais escrutínio, e não menos, precisamente porque frequentemente entram no comércio antes de sua farmacologia estar mapeada em humanos. O atalho antigo era perguntar se a molécula se liga a CB1 ou CB2. A pergunta melhor é o que mais ela faz e se essas ações se tornam relevantes em doses reais após inalação, ingestão oral ou metabolismo.
Para dor, canais TRP e canais de sódio são exemplos óbvios. David Julius e Ardem Patapoutian receberam o Prêmio Nobel de Fisiologia ou Medicina de 2021 por descobertas de receptores para temperatura e toque, lembrando que a biologia somatossensorial é construída sobre alvos que os cannabinoid podem influenciar fora dos receptores cannabinoid clássicos. A ativação de TRPV1 pode contribuir para analgesia por dessensibilização, mas também pode produzir irritação e é fortemente dependente da concentração. Um regulador que olhe apenas atividade em CB1 vai perder todo esse eixo risco-benefício.
Para segurança psiquiátrica, a sinalização serotoninérgica importa. O CBD foi repetidamente discutido como candidato ansiolítico ligado a 5-HT1A, mas o envolvimento de serotonina pode ser indireto e dependente do contexto. Essa incerteza não é motivo para ignorar o alvo; é motivo para estudá-lo com cuidado antes que os produtos sejam normalizados como inofensivos. O mesmo vale para GPR55, às vezes apresentado como candidato a “CB3” apesar do debate em andamento. Um alvo contestado ainda é um alvo que pode moldar sinalização de cálcio, excitabilidade e respostas inflamatórias.
Para efeitos metabólicos e inflamatórios, alvos intracelulares como PPAR-gamma complicam ainda mais o quadro. A sinalização por receptores nucleares não parece um efeito rápido de intoxicação, mas pode importar para exposição crônica, distribuição no tecido adiposo, alterações transcricionais e interação com outros estados de doença. Mensagens de saúde pública baseadas em se algo “dá high” perdem esses riscos lentos e silenciosos.
É aqui que regulação, toxicologia e comunicação de segurança ao consumidor precisam de alfabetização em alvos moleculares. Nem todo acerto in vitro terá relevância clínica. Diferenças entre espécies são reais. Metabólitos podem dominar. Mas as agências devem exigir painéis de alvo, caracterização de metabólitos, dados de concentração-resposta e estudos humanos relevantes para comprometimento antes de presumir que um novo cannabinoid pode ser governado como outro qualquer. O artigo de 2016 do ACS Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, reflete como a descoberta de fármacos já funciona hoje: desenho estruturado, engenharia de seletividade e atenção explícita a diferenças em nível de receptor. A regulação deveria parar de fingir que o mercado é mais simples do que a própria química medicinal já sabe que ele é.
A saúde pública não pode se dar ao luxo do reducionismo de receptor. “Cannabinoid” é um rótulo de partida. Não é uma conclusão de segurança.
Descoberta de fármacos: projetando cannabinoid e moléculas inspiradas em cannabinoid para alvos não CB1/CB2
A descoberta de fármacos em torno dos cannabinoid já foi muito além da velha pergunta sobre se uma molécula é “ativa em CB1” ou “ativa em CB2.” Essa simplificação sempre foi instável, porque muitos compostos relacionados a cannabinoid são farmacologicamente confusos: atingem canais iônicos, GPCR fora do par canônico, receptores nucleares intracelulares, processos de transporte e enzimas de metabolização, frequentemente com efeitos diferentes conforme concentração, tecido e via de administração. Para químicos medicinais, essa confusão não é apenas um problema. É também uma oportunidade.
O objetivo central de design é claro o suficiente: manter efeitos analgésicos, anti-inflamatórios ou ansiolíticos, enquanto reduz riscos ligados à forte ativação de CB1 no cérebro. Esses riscos não são abstratos. Sedação, comprometimento cognitivo, intoxicação, potencial de abuso e efeitos psiquiátricos limitantes de dose são exatamente por que “THC-like” costuma ser um perfil ruim em um programa de desenvolvimento, mesmo quando o próprio THC mostra farmacologia útil. O clima regulatório atual reforça esse ponto. Em 2025, o HHS apoiou ação de agendamento ao afirmar que “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety”, lembrando que intoxicantes modificados ou intensificados não podem ser assumidos como se se comportassem como compostos vegetais familiares apenas porque estão próximos na prateleira de marketing. A farmacologia em nível de alvo importa.
Restrição periférica, seletividade funcional e sinalização enviesada
Uma rota para evitar efeitos adversos centrais é direta, embora eficaz: manter o fármaco fora do cérebro. A restrição periférica pode ser projetada aumentando a área de superfície polar, a capacidade de ligação por hidrogênio, ajustando pKa ou tornando a molécula substrato de transportadores de efluxo na barreira hematoencefálica. A ideia não é exclusiva da ciência cannabinoid, mas se encaixa muito bem nela porque a sinalização da dor frequentemente começa em nociceptores periféricos, tecidos inflamados e gânglios da raiz dorsal.
É aí que alvos fora de CB1/CB2 se tornam especialmente atraentes. Um relatório de 2025 da Hebrew University argumentou que THC inibe nociceptores periféricos ao atingir canais de sódio NaV1.7 e NaV1.8, dois dos principais motores da excitabilidade nociceptiva. Se esse mecanismo se mantiver em diferentes sistemas, isso importará muito. NaV1.7 há muito é tratado como alvo premium para dor porque mutações humanas de perda de função em SCN9A podem produzir insensibilidade congênita profunda à dor. Um scaffold cannabinoid que preserve modulação de canais de sódio na periferia enquanto minimiza a sinalização central de CB1 não seria apenas “THC menos intoxicante”. Seria um tipo diferente de analgésico.
Os canais TRP criam uma abertura semelhante. A biologia sensorial mais ampla aqui foi reconhecida no nível mais alto quando o Prêmio Nobel de Fisiologia ou Medicina de 2021 foi concedido a David Julius e Ardem Patapoutian por descobertas de receptores para temperatura e toque. TRPV1, TRPA1 e canais relacionados estão profundamente ligados à nocicepção e à sinalização inflamatória, e vários phytocannabinoid interagem com eles de maneiras sensíveis à concentração e, às vezes, paradoxais: a ativação inicial pode ser seguida por dessensibilização, contribuindo potencialmente para analgesia. Isso torna a química medicinal mais difícil, não mais fácil. Mas também significa que um composto não precisa ser um ligante ortostérico limpo de receptor CB para ter valor terapêutico.
A seletividade funcional acrescenta outra camada. Mesmo em CB1 ou CB2, ligantes podem favorecer uma saída de sinalização sobre outra, deslocando o equilíbrio entre vias de proteína G, recrutamento de beta-arrestina, internalização do receptor e programas transcricionais a jusante. Em termos simples, duas moléculas podem ambas “ligar CB1” e ainda assim se comportar muito diferente em tecido vivo. O artigo de 2016 do Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, reflete o quanto a estratégia de design se deslocou para ajuste baseado em estrutura em vez de rótulos grosseiros de receptor. A mesma lógica agora vai além dos receptores CB: os químicos querem scaffolds cuja forma, lipofilicidade e restrições conformacionais os enviesem para um mecanismo relevante para dor ou ansiedade, evitando a bagagem pesada da sinalização central.
O CBD é a prova permanente de que um fármaco relacionado a cannabinoid pode importar clinicamente sem ser explicado por agonismo de CB1/CB2. A rotulagem da FDA para solução oral de cannabidiol, atualizada em 2024, cobre convulsões associadas à síndrome de Lennox-Gastaut, à síndrome de Dravet e ao complexo de esclerose tuberosa em pacientes com 1 ano de idade ou mais. Qualquer que seja a combinação de efeitos em TRPV1, 5-HT1A, GPR55, adenosina, intracelular e de nível de rede que finalmente explique essa eficácia, não é uma história simples de CB1. Os pesquisadores perceberam isso.
MIRA-55 e a busca por mecanismos diferenciados
MIRA-55 é um caso útil não porque encerre qualquer coisa, mas porque mostra como as empresas agora enquadram programas cannabinoid. Em um comunicado de 2025 veiculado pela Nasdaq, a MIRA Pharmaceuticals disse que seu candidato mostrava um “differentiated mechanism of action” e “anxiolytic activity relative to THC” em trabalhos pré-clínicos. Essa formulação diz quase tudo sobre o sinal atual para investidores e reguladores. Ser inspirado em cannabinoid já não basta. A empresa quer distância da imitação simples de THC, especialmente para indicações como ansiedade, nas quais efeitos adversos centrais podem apagar o benefício.
Ainda assim, ceticismo é obrigatório. “Differentiated mechanism” em um comunicado corporativo é uma alegação, não uma conclusão. Pode significar engajamento alterado de receptor, distribuição tecidual diferente, metabólitos distintos, agonismo parcial, viés funcional, efeitos fora do alvo em canal iônico ou simplesmente um perfil comportamental diferente em um único ensaio com animal. Sem painéis completos de farmacologia, curvas de concentração-resposta, identificação de metabólitos, dados de ocupação de receptor e replicação cega, a expressão é mais hipótese do que resultado.
Dito isso, a estratégia por trás da alegação é plausível. Se um composto consegue reduzir comportamento semelhante à ansiedade enquanto diminui intoxicação, alteração de memória ou supressão locomotora em relação ao THC, a química medicinal provavelmente mudou uma ou mais de quatro coisas: penetração cerebral, eficácia intrínseca em CB1, engajamento de alvos fora de CB como 5-HT1A ou canais TRP, ou conversão metabólica em espécies ativas com perfil-alvo diferente. Esses são exatamente os eixos em que os programas cannabinoid modernos competem.
MIRA-55 também ilustra o problema de deconvolução de alvos. Moléculas semelhantes a cannabinoid costumam ser “dirty” no sentido farmacológico. Isso não é um julgamento moral; é um alerta mecanístico. Se surge um sinal ansiolítico pré-clínico, não se pode presumir que um único receptor o explique. A sinalização serotoninérgica pode ser direta ou indireta. Os efeitos em PPAR-gamma podem exigir acúmulo intracelular ou metabólitos. GPR55 pode parecer importante em um ensaio e marginal em outro. Um acerto in vitro a 10 micromolar pode ser irrelevante se as concentrações livres no cérebro nunca se aproximarem disso in vivo.
Como é um pipeline realista de cannabinoid
Um pipeline realista é mais estreito e mais disciplinado do que a retórica pública sugere. Não é uma parada de “compostos de cannabis não psicoativos” correndo rumo à aprovação. É um filtro.
Na frente estão a modificação de scaffold e a triagem: cannabinoid clássicos, cannabinoid não clássicos, lipídios inspirados em endocannabinoid e quimiotipos não relacionados que imitam uma característica útil da farmacologia cannabinoid sem herdar o pacote inteiro. Os químicos alteram comprimento da cadeia lateral, restrições de anel, estereoquímica, posicionamento de heteroátomos e pontos fracos metabólicos, depois testam não apenas CB1 e CB2, mas TRPV1, TRPA1, canais NaV, GPCR órfãos selecionados e ensaios relevantes para serotonina. Os acertos então enfrentam estudos de ADME, análise de concentração não ligada e medições cérebro/plasma. Muitos morrem ali.
Os programas que sobrevivem normalmente se dividem em algumas categorias críveis. Uma é a dos analgésicos com restrição periférica, onde o sonho é alívio da dor via canais de sódio, dessensibilização de TRP, modulação inflamatória ou sinalização cannabinoid periférica sem exposição importante ao SNC. Outra é a dos ligantes CB1 enviesados ou de baixa eficácia, que buscam preservar a sinalização terapêutica enquanto reduzem a intoxicação. Uma terceira é a dos compostos de mecanismo misto, muitas vezes mais realistas do que o purismo de alvo único, em que ações modestas em vários nós de dor ou ansiedade superam um ligante “limpo”, porém clinicamente fraco.
O gancho translacional que mantém essa área viva é simples: parece possível separar analgesia do high, pelo menos em sistemas em estágio de pesquisa. O resumo da ScienceDaily de 2026 sobre “a cannabis compound that relieves pain without the high” deve ser tratado com cuidado, porque manchetes correm à frente dos dados, mas o conceito se encaixa na direção geral da química medicinal. O mesmo vale para o trabalho sobre canais de sódio em THC. O mesmo vale para esforços de construir ligantes seletivos por docking e design estrutural. O padrão é consistente mesmo quando alegações individuais precisam de ajustes.
O que o pipeline provavelmente não parece é sucesso clínico massivo no curto prazo. Promiscuidade de alvo, complexidade de metabólitos, diferenças entre espécies e efeitos de formulação continuam quebrando histórias simples. Mas o campo já não está preso à pergunta sobre se os cannabinoid são “realmente” sobre CB1 e CB2. Os desenvolvedores de fármacos já responderam a isso com seus orçamentos de química. Eles estão projetando para o espaço além desses receptores, porque é lá que hoje está a melhor chance de separar benefício de risco.
Equívocos comuns e controvérsias não resolvidas
O maior erro na discussão pública sobre cannabinoid é o reducionismo de receptor: a vontade de forçar uma farmacologia bagunçada em um alvo de manchete. Esse hábito sempre foi frágil, e está ficando mais arriscado à medida que reguladores e desenvolvedores de fármacos enfrentam compostos que já não são apenas “THC” ou “CBD” no sentido simples, derivado de planta. Em 2025, o HHS disse que “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” ao apoiar a ação da DEA contra produtos intensificados com 7-OH. Classe de droga diferente, mesma lição: quando os químicos alteram, enriquecem ou semissintetizam um scaffold intoxicante, velhas suposições sobre ação em receptor e segurança podem falhar rapidamente. A ciência da cannabis tem o mesmo problema. Um cannabinoid não é uma chave mágica para uma única fechadura. É, em geral, um ligante promíscuo cujos efeitos no mundo real dependem da concentração, da exposição tecidual, do metabolismo e dos alvos fora do alvo que se tornam relevantes nesses níveis.
Existe mesmo um receptor CB3
Não existe um receptor CB3 consensual.
Essa resposta parece direta porque as evidências exigem isso. Vários receptores foram propostos ao longo dos anos como candidatos a “CB3”, especialmente GPR55, às vezes GPR18 e, ocasionalmente, outros GPCR órfãos que respondem em alguns sistemas de ensaio a ligantes cannabinoid ou lipídios relacionados ao endocannabinoid. Mas um alvo proposto não é uma classe de receptor aceita. CB1 e CB2 ganharam seus nomes por meio de evidências convergentes: clonagem, farmacologia de ligantes reproduzível, distribuição tecidual, sinalização e ampla replicação. Os candidatos a CB3 não passaram nesse teste.
GPR55 é o suspeito usual. Ele é expresso no cérebro, em células imunes, no intestino e no osso, e alguns cannabinoid interagem com ele. O CBD costuma ser descrito como antagonista de GPR55 em ensaios celulares; certos cannabinoid sintéticos também mostram atividade. Ainda assim, a farmacologia é inconsistente entre laboratórios, ligantes e leituras. Alguns compostos parecem ativos em um ensaio de sinalização e silenciosos em outro. Diferenças entre espécies complicam o quadro. Os ligantes endógenos são debatidos. Mais importante: chamar GPR55 de “CB3” sugere um lugar resolvido dentro da família canônica de receptores cannabinoid que o campo ainda não lhe concedeu.
Isso importa porque rótulos podem correr mais rápido que as evidências. Quando um receptor ganha um apelido chamativo, o apelido passa a fazer o trabalho explicativo que ainda não conquistou. Dor? CB3. Ansiedade? CB3. Efeitos ósseos? CB3. Isso não é farmacologia; é branding. A posição mais cuidadosa é que GPR55, GPR18, GPR119 e receptores relacionados são alvos fora de CB1/CB2 com graus variados de evidência para modulação por cannabinoid ou lipídios semelhantes a cannabinoid. Alguns podem importar muito em tecidos específicos. Nenhum alcançou status consensual como “o terceiro receptor cannabinoid”.
A descoberta de fármacos se deslocou de acordo. Artigos de química medicinal não agem como se um novo nome de receptor resolvesse o sistema. O artigo de 2016 do Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, é um bom marcador de onde o campo realmente está: design baseado em estrutura, seletividade de receptor, engenharia de scaffold e otimização guiada por alvo, não mitologia sobre um misterioso CB3 esperando explicar tudo. Da mesma forma, alegações de empresas sobre compostos cannabinoid de próxima geração agora frequentemente enfatizam mecanismos diferenciados, em vez de apenas agonismo de receptor mais forte. O comunicado pré-clínico de 2025 da MIRA Pharmaceuticals para o MIRA-55 afirmava explicitamente um “differentiated mechanism of action” e atividade ansiolítica relativa ao THC. Isso é material promocional, não ciência resolvida, mas reflete uma mudança estratégica real: terapêuticos cannabinoid úteis podem vir de se afastar da intoxicação bruta de CB1, e não de descobrir um receptor que explique tudo.
O CBD funciona principalmente por serotonina
Também não, embora a sinalização de serotonina faça parte da história.
O CBD é frequentemente comercializado na cultura popular como se fosse basicamente um fármaco natural de 5-HT1A. Essa simplificação persiste porque há evidência real por trás dela. Em estudos pré-clínicos, o CBD mostrou efeitos semelhantes a ansiolíticos e antiestresse que, em alguns paradigmas, são reduzidos por antagonistas de 5-HT1A. Estudos experimentais em humanos também sugeriram que mecanismos serotoninérgicos podem contribuir para efeitos ansiolíticos agudos em certas condições. Mas “contribui para” não é “funciona principalmente por”.
O CBD é farmacologicamente amplo. Tem baixa afinidade por CB1 e CB2 em comparação com THC, mas isso não o transforma automaticamente em um agente serotoninérgico de alvo único. Ao longo da literatura, contribuintes plausíveis incluem TRPV1, 5-HT1A, GPR55, sinalização de adenosina, PPAR-gamma, manejo intracelular de cálcio, tônus do endocannabinoid relacionado à FAAH em alguns contextos e efeitos de metabólitos que podem não corresponder ao composto original. A concentração importa aqui mais do que muitos explicadores admitem. Um efeito em receptor observado in vitro em nível micromolar pode não dominar em humanos após uma dose oral padrão, em que a absorção é variável e o metabolismo de primeira passagem é substancial.
O registro clínico vai contra alegações de mecanismo único. O uso mais reconhecido pela FDA de CBD purificado não é ansiedade, mas distúrbios convulsivos: a solução oral é indicada para síndrome de Lennox-Gastaut, síndrome de Dravet e complexo de esclerose tuberosa em pacientes com 1 ano ou mais. Esse uso aprovado já diz algo. Se o CBD fosse “principalmente serotonina”, seu perfil terapêutico mais reprodutível seria difícil de conciliar com o que os clínicos realmente usam. A serotonina pode importar em contextos relacionados à ansiedade, especialmente respostas ligadas a 5-HT1A, mas a farmacologia humana do CBD não se reduz a um rótulo serotoninérgico.
Mesmo na ansiedade, o mecanismo provavelmente muda com dose e contexto. TRPV1 é um bom exemplo. O CBD pode ativar TRPV1, e a sinalização TRP é central o suficiente para a biologia sensorial que David Julius e Ardem Patapoutian receberam o Nobel de 2021 por descobertas de receptores para temperatura e toque. Ainda assim, os efeitos de TRPV1 não são lineares. A ativação pode ser seguida por dessensibilização; doses baixas e altas podem produzir resultados comportamentais diferentes. Então, quando alguém diz “CBD funciona por serotonina”, a resposta correta é: em parte, às vezes, e provavelmente não sozinho.
Um único alvo pode explicar um efeito de planta inteira
Nenhum alvo único pode explicar um efeito de cannabis de planta inteira, e tentar forçá-lo normalmente obscurece mais do que esclarece.
Os efeitos de planta inteira surgem de variáveis empilhadas. Comece pela composição: THC, CBD, minor cannabinoids como CBG, CBC, THCV, precursores ácidos, produtos de oxidação e metabólitos, todos trazem perfis-alvo distintos. Acrescente a via de administração e o quadro muda de novo. Cannabinoid inalados chegam rapidamente ao cérebro; produtos orais passam por metabolismo de primeira passagem e geram espécies ativas diferentes. A lei dos EUA ainda define hemp por um limite de delta-9 THC de “not more than 0.3 percent on a dry weight basis”, mas essa linha legal diz pouco sobre a farmacologia de todo o resto da amostra ou sobre o que se forma após o metabolismo.
Depois adicione especificidade tecidual. Um cannabinoid pode afetar CB1 central, canais TRP periféricos, sinalização imune e receptores nucleares em paralelo. O relatório de 2025 da Hebrew University de que THC inibe nociceptores periféricos ao atingir NaV1.7 e NaV1.8 é um exemplo forte porque quebra a equação preguiçosa “efeito de THC=efeito de CB1”. Se até o THC tem ações significativas em canais de sódio nas vias da dor, a ideia de que uma flor, extrato ou comestível tem um receptor mestre fica difícil de defender. A pesquisa destacada pela ScienceDaily em 2026 — um cannabinoid que alivia a dor sem o high — aponta na mesma direção, embora ainda em estágio de pesquisa. A analgesia pode ser separada da intoxicação central explorando restrição periférica ou alvos fora de CB1. É para isso que o campo está caminhando.
Expectativa e biologia individual também importam. Experiência prévia, nível de ansiedade, genética, sexo, atividade de enzimas hepáticas, sono, inflamação e medicamentos concomitantes mudam o que um determinado produto parece e o que ele faz fisiologicamente. Terpenes podem contribuir em alguns casos, mas não devem ser tratados como condutores mágicos de todo o efeito. Suas concentrações costumam ser baixas, e as evidências humanas são muito mais frágeis do que a linguagem de marketing sugere.
A visão mais dura, porém mais precisa, é esta: os efeitos da cannabis são emergentes. Eles surgem de muitas interações modestas, não de um receptor-slogan. Isso torna a ciência menos arrumada. Também a torna mais honesta.
Interpretação prática para leitores, clínicos e pesquisadores
A lição prática da farmacologia cannabinoid fora de CB1/CB2 é simples, porém exigente: alegações de mecanismo devem receber escrutínio mais duro, não aceitação mais fácil, quando um artigo invoca canais TRP, PPARs, GPR55, 5-HT1A, sinalização da adenosina ou canais NaV. Cannabinoid são frequentemente moléculas farmacologicamente promíscuas. Isso pode ser útil na descoberta de fármacos. Também pode induzir leitores ao erro, fazendo-os tratar qualquer acerto em receptor em uma placa como explicação para um efeito clínico.
Uma boa regra é classificar as evidências pela distância até o paciente. Um ensaio de ligação é o começo, não o fim. Estudos de sinalização celular vêm em seguida. Trabalho com animais pode afiar a plausibilidade. A farmacologia experimental em humanos importa mais. Evidência de medicamento aprovado importa mais de tudo, e mesmo aí o rótulo pode não resolver o mecanismo. A solução oral de cannabidiol, por exemplo, é aprovada pela FDA para convulsões associadas à síndrome de Lennox-Gastaut, à síndrome de Dravet e ao complexo de esclerose tuberosa em pacientes com 1 ano ou mais, mas seu perfil terapêutico não é explicado de forma limpa por ativação de CB1 ou CB2. Essa lacuna é exatamente o motivo pelo qual as alegações sobre TRPV1, GPR55, 5-HT1A e alvos intracelulares continuam reaparecendo.
Como ler alegações de mecanismo cannabinoid criticamente
Comece com a espécie e o sistema. O efeito foi mostrado em tecido humano, linha celular de roedor, ovócitos de Xenopus ou receptores superexpressos em células HEK293? Isso não é intercambiável. GPR55 é um exemplo clássico: um estudo pode mostrar o CBD como antagonista em um sistema recombinante, enquanto outro contexto produz sinalização fraca ou variável porque expressão do receptor, lipídios endógenos e desenho do ensaio diferem. Chamar isso de “o mecanismo” costuma ser prematuro.
Depois pergunte sobre a concentração. É aqui que muitas alegações sobre cannabinoid desmoronam. Um artigo pode relatar ativação de TRPV1, transativação de PPAR-gamma ou inibição de canais de sódio em concentrações micromolares. Ótimo. Mas a dose humana discutida realmente produz concentrações livres em tecido nessa faixa? Cannabinoid orais enfrentam metabolismo de primeira passagem, ligação extensa a proteínas e distribuição tecidual irregular. Um acerto de alvo em 30 micromolar in vitro pode ser química interessante e farmacologia irrelevante à beira do leito. O inverso também pode acontecer: acúmulo local em tecido, metabólitos ativos ou partição lipídica podem tornar um alvo intracelular mais plausível do que os níveis plasmáticos sozinhos sugerem. De qualquer forma, concentração não é um detalhe lateral. É o detalhe.
A medição direta do alvo também importa. Os investigadores realmente bloquearam o efeito com um antagonista seletivo, reduziram a expressão do receptor ou mediram corrente de canal? Ou inferiram o mecanismo por semelhança com a literatura anterior? Para canais TRP, especialmente TRPV1 e TRPA1, isso importa porque a ativação pode ser bifásica e dessensibilizante. Um composto pode primeiro ativar um canal e depois reduzir a responsividade a jusante, o que significa que “agonista” nem sempre se traduz limpidamente em “mais dor” ou “mais sensação de calor”. Isso é uma das razões pelas quais o Nobel de 2021 para David Julius e Ardem Patapoutian, concedido “for their discoveries of receptors for temperature and touch,” é tão relevante aqui: a sinalização somatossensorial é mecanisticamente rica, e rótulos simplistas de receptor frequentemente falham.
A história NaV mostra o que evidência mais forte parece. Pesquisadores da Hebrew University relataram em 2025 que THC inibe nociceptores periféricos ao atingir canais de sódio nociceptivos NaV1.7 e NaV1.8. Essa alegação é mais informativa do que afirmações vagas de que THC “atua fora de CB1”, porque nomeia canais relevantes para a dor já centrais na pesquisa de analgesia. Também reformula uma suposição antiga. Até mesmo um composto famoso pela intoxicação mediada por CB1 pode ter ações não CB clinicamente relevantes, especialmente em tecido periférico.
Os leitores também devem observar explicações concorrentes. Suponha que um estudo ligue o CBD à ansiolise por meio de 5-HT1A. Hipótese razoável. Mas foi descartada a sedação? A modulação de CB1 foi indireta? O experimento distinguiu efeitos em nível de receptor de mudanças no tônus do endocannabinoid, captação de adenosina, sinalização inflamatória ou expectativa em sujeitos humanos? Fármacos de múltiplos alvos raramente anunciam qual alvo está fazendo a maior parte do trabalho em qualquer modelo.
O clima regulatório atual torna esse ceticismo mais do que acadêmico. Em 2025, o HHS afirmou que “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” ao apoiar ação regulatória sobre produtos intensificados de 7-OH. Isso não é um caso de cannabis, mas a lição se transfere com clareza: os reguladores distinguem cada vez mais constituintes vegetais familiares de intoxicantes intensificados, semissintéticos ou de outra forma modificados, com diferentes potências e perfis de segurança. Política sobre cannabinoid que trata todos os compostos como versões escaladas de delta-9-THC está farmacologicamente ultrapassada.
Perguntas que os clínicos devem fazer sobre relevância de alvo
Clínicos não precisam memorizar todos os GPCR órfãos para interpretar bem alegações sobre cannabinoid. Eles precisam de uma lista de verificação disciplinada.
Primeiro: o efeito foi mostrado em humanos ou apenas em células e animais? Um modelo inflamatório em camundongo pode sustentar plausibilidade para envolvimento de PPAR-gamma ou TRPA1, mas não estabelece benefício para o paciente. Segundo: em quais concentrações ou doses? Se um alvo proposto é engajado apenas em níveis acima do que a dose oral padrão atinge, esse mecanismo talvez não explique os resultados clínicos de rotina. Terceiro: o alvo foi medido diretamente? Ocupação de receptor, eletrofisiologia, reversão por antagonista ou perturbação genética contam mais do que inferência narrativa.
Quarto: a via de administração torna a alegação mais ou menos crível? Inalação, ingestão oral, administração transdérmica e aplicação tópica criam perfis de exposição muito diferentes. Um mecanismo analgésico periférico é mais plausível para um composto tópico ou perifericamente restrito do que para uma molécula que rapidamente inunda o cérebro. Essa distinção importa se o objetivo terapêutico é analgesia sem intoxicação.
Quinto: há explicações concorrentes ligadas a efeitos adversos conhecidos? Se um paciente relata menos ansiedade após uma preparação cannabinoid, isso é um efeito ansiolítico mediado por 5-HT1A, menos dor, sedação inespecífica ou expectativa? Se marcadores inflamatórios mudam, PPAR-gamma é o provável motor, ou ocorreram mudanças metabólicas ou imunes mais amplas a montante? Mecanismo não deve ser inferido apenas pela melhora de sintomas.
Para clínicos, minor cannabinoids merecem a mesma cautela. CBC, CBG, THCV e cannabinoid ácidos costumam ser discutidos como se cada um viesse com um perfil-alvo estável. A literatura ainda não justifica essa confiança. Alguns são promissores. Nenhum deve ser tratado como farmacologicamente resolvido só porque um nome de marca ou uma discussão em rede social afirma especificidade de receptor.
Para onde o campo provavelmente vai a seguir
A direção mais crível no curto prazo é a analgesia periférica. O apelo translacional é óbvio: separar alívio da dor da intoxicação central. O resumo de 2026 da ScienceDaily descrevendo trabalho sobre “a cannabis compound that relieves pain without the high” deve ser lido como sinal em estágio de pesquisa, não como resposta clínica final, mas captura a direção que a química medicinal está buscando. O mesmo vale para o trabalho de 2025 da Hebrew University sobre NaV1.7/NaV1.8: a biologia da dor está empurrando a ciência cannabinoid para nervos periféricos, canais iônicos e exposição tecidual seletiva.
Ligantes de receptor nuclear anti-inflamatórios são outra via forte. Alegações sobre PPAR-gamma precisam de melhores dados de engajamento de alvo em humanos, mas o conceito é plausível o suficiente para justificar desenvolvimento sério, especialmente onde vias metabólicas e inflamatórias se cruzam. Ansiolíticos de múltiplos alvos também provavelmente continuarão sendo um território ativo. Em um comunicado da Nasdaq de 2025, a MIRA Pharmaceuticals disse que seu candidato MIRA-55 mostrou um “differentiated mechanism of action” e “anxiolytic activity relative to THC” em dados pré-clínicos. Como isso é reportado pela empresa e em nível pré-clínico, deve ser tratado com cautela. Ainda assim, reflete uma tendência real: pesquisadores já não estão satisfeitos em perguntar se um candidato é “tipo THC” ou “tipo CBD.” Eles querem perfis-alvo definidos.
A mesma mudança é visível na química medicinal. O artigo de 2016 do Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, sinalizou uma abordagem estrutural que só cresceu desde então: construir ligantes para alvos específicos, tecidos específicos e vieses de sinalização específicos. Minor cannabinoids mais bem definidos provavelmente surgirão dessa mentalidade, e não de alegações amplas de que todo cannabinoid raro tem um nicho de bem-estar único.
A lição prática é direta. Ao ler farmacologia cannabinoid, faça sempre cinco perguntas: O efeito foi mostrado em humanos ou apenas em células? Em quais concentrações? O alvo foi medido diretamente? A dose alcança esse alvo em tecido vivo? Há explicações concorrentes? O futuro da ciência cannabinoid não é uma farmacologia menos específica, mas mais.
Referências
- [1]HHS and FDA support DEA action on dangerous 7-OH products. HHS Press Room, 2025. https://www.hhs.gov/press-room/hhs-fda-support-dea-7-oh-scheduling.html
- [2]EPIDIOLEX (cannabidiol) oral solution label. FDA drug label, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/210365s016lbl.pdf
- [3]Library Docking for Cannabinoid-2 Receptor Ligands. Journal of Medicinal Chemistry, 2016. https://pubs.acs.org/doi/10.1021/acs.jmedchem.6c00835
- [4]MIRA Pharmaceuticals Reports New Preclinical Data Demonstrating MIRA-55's Differentiated Mechanism of Action and Anxiolytic Activity Relative to THC. Nasdaq press release, 2025. https://www.nasdaq.com/press-release/mira-pharmaceuticals-reports-new-preclinical-data-demonstrating-mira-55s
- [5]Psychoactive cannabinoid THC inhibits peripheral nociceptors by targeting NaV1.7 and NaV1.8 nociceptive sodium channels. Research portal summary, 2025. https://cannabinoids.huji.ac.il/publications/psychoactive-cannabinoid-thc-inhibits-peripheral-nociceptors-targeting
- [6]A cannabis compound that relieves pain without the high. ScienceDaily, 2026. https://www.sciencedaily.com/releases/2026/06/260619033343.htm
- [7]The Nobel Prize in Physiology or Medicine 2021. Nobel Prize Press Release, 2021. https://www.nobelprize.org/prizes/medicine/2021/press-release/
- [8]Agriculture Improvement Act of 2018. Congress.gov, 2018. https://www.congress.gov/bill/115th-congress/house-bill/2/text