






Table des matières
- Pourquoi la science des cannabinoid ne peut pas se réduire à CB1 et CB2
- Le système endocannabinoïde face au paysage plus large des cibles des cannabinoid
- Canaux TRP : les capteurs de chaleur, de douleur et d’irritation que les cannabinoid continuent d’atteindre
- PPAR : les cannabinoid comme signaux lipidiques intracellulaires, et non comme de simples ligands de récepteurs membranaires
- GPR55, GPR18 et GPR119 : le problème des GPCR orphelins
- Signalisation de la sérotonine : là où les cannabinoid interfèrent avec les systèmes 5-HT
- Au-delà de la liste demandée : les canaux sodiques et d’autres cibles non canoniques qui modifient déjà la conversation sur la douleur
- En quoi les cannabinoid spécifiques diffèrent lorsque l’on ne s’interroge plus seulement sur CB1 et CB2
- La méthode compte : pourquoi la conception des essais façonne ce que nous pensons que font les cannabinoid
- Niveaux de preuve : de la boîte de culture au clinic
- Sécurité, réglementation et pourquoi la pharmacologie hors cible compte en santé publique
- Découverte de médicaments : concevoir des cannabinoid et des molécules inspirées des cannabinoid pour des cibles autres que CB1/CB2
- Idées reçues courantes et controverses non résolues
- Interprétation pratique pour les lecteurs, les cliniciens et les chercheurs
Pourquoi la science des cannabinoid ne peut pas se réduire à CB1 et CB2
La version simplifiée de la pharmacologie des cannabinoid dit ceci : THC agit sur CB1, les effets immunitaires passent par CB2, et tout le reste n’est qu’une note de bas de page. Cette formulation est facile à enseigner et facile à répéter. Elle est aussi souvent fausse, au point d’empêcher une compréhension sérieuse de la douleur, de l’inflammation, de l’anxiété, du prurit, des nausées, du métabolisme et de la neuroprotection.
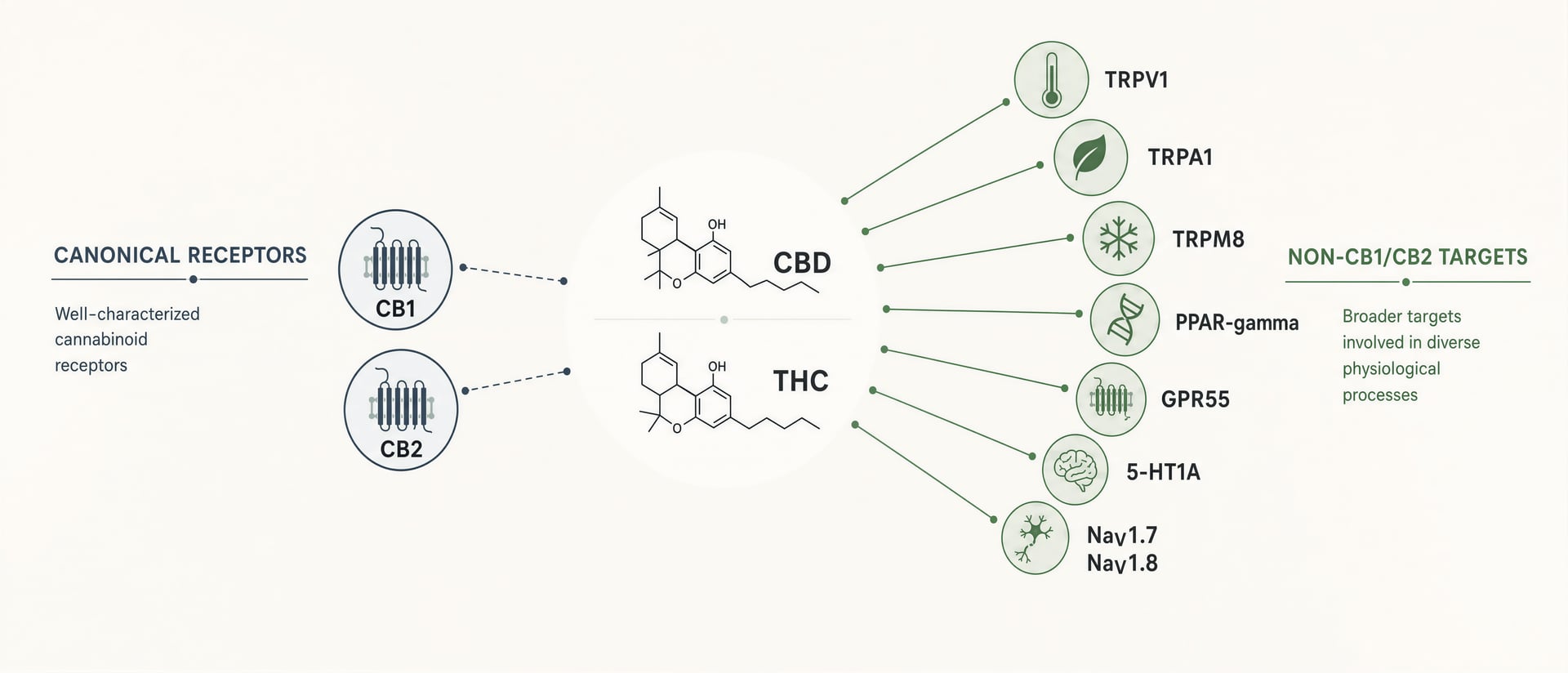
CB1 et CB2 comptent. CB1 est abondant dans le cerveau et explique en grande partie l’intoxication induite par le THC, les troubles de la mémoire, les effets sur l’appétit et une partie de son action analgésique. CB2 est central dans de nombreuses discussions sur l’immunité et l’inflammation. Mais les cannabinoid ne sont pas des ligands propres, conçus pour un seul récepteur chacun. Ce sont des molécules lipophiles, flexibles sur le plan conformationnel, qui interagissent avec un champ pharmacologique plus vaste : les canaux transitoires de potentiel récepteur tels que TRPV1 et TRPA1, les récepteurs nucléaires tels que PPAR-gamma, les GPCR orphelins ou encore débattus proches des cannabinoid tels que GPR55 et GPR18, les récepteurs de la sérotonine dont 5-HT1A, la signalisation liée à l’adénosine, le transport et le métabolisme des acides gras, et, dans des travaux plus récents, les canaux sodiques voltage-dépendants, notamment NaV1.7 et NaV1.8.[1]HHS and FDA support DEA action on dangerous 7-OH products. U.S. Department of Health and Human Services. HHS Press Room, 2025. https://www.hhs.gov/press-room/hhs-fda-support-dea-7-oh-scheduling.html
Ce champ élargi est important parce que le mécanisme détermine le risque et le bénéfice. Les régulateurs sont déjà confrontés à ce problème dans des débats voisins sur les politiques en matière de drogues. En 2025, le Department of Health and Human Services des États-Unis a déclaré que « 7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety » en soutenant l’action de la DEA sur les produits à 7-OH renforcé. Ce composé n’est pas un cannabinoid, mais la leçon politique est transposable : dès que des chimistes commencent à modifier des squelettes de produits naturels et à concentrer des métabolites, les catégories simples fondées sur la source cessent de protéger le public. Le profil cible d’une molécule compte davantage que la manière dont les écrits populaires la présentent comme familière.
Le mythe du récepteur dans les textes populaires sur le cannabis
Les explications populaires sur le cannabis présentent généralement les récepteurs comme des interrupteurs marche/arrêt : THC active CB1, CBD ne « se lie pas fortement », donc CBD doit être faible ou mystérieux. Cette version confond plusieurs idées pharmacologiques distinctes en un seul verbe vague, « se lier ».
L’agonisme orthostérique est le cas classique. Un ligand occupe le site actif principal du récepteur et stabilise la signalisation. THC est un agoniste partiel de CB1 et CB2. Il s’agit d’un mode d’action parmi d’autres, pas d’un modèle unique pour toute la biologie des cannabinoid. Une molécule peut aussi agir allostériquement, en modifiant l’action d’un autre ligand sur le récepteur sans occuper le même site. Elle peut ouvrir, sensibiliser ou désensibiliser un canal ionique. Elle peut pénétrer dans la cellule et activer un récepteur nucléaire qui modifie la transcription génique sur des heures plutôt que sur des millisecondes. Elle peut inhiber un transporteur, modifier les propriétés de la membrane, ou ralentir une enzyme qui dégrade un lipide de signalisation endogène.[2]EPIDIOLEX (cannabidiol) oral solution label. U.S. Food and Drug Administration. FDA drug label, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/210365s016lbl.pdf
CBD est la réfutation la plus claire du réductionnisme récepteur. Son usage clinique approuvé ne repose pas sur un agonisme de CB1. La notice de la solution orale de cannabidiol de la FDA indique qu’elle est destinée aux crises associées au syndrome de Lennox-Gastaut, au syndrome de Dravet et au complexe de sclérose tubéreuse chez les patients âgés d’un an et plus. Quel que soit le mécanisme complet qui sous-tend cet effet, il n’est pas correctement expliqué par l’ancienne idée selon laquelle une action cannabinoid significative équivaut à une forte activation de CB1 ou CB2. Parmi les candidats mécanistiques fréquemment évoqués dans la littérature figurent TRPV1, une signalisation liée à 5-HT1A, la modulation de l’adénosine, des effets sur le calcium intracellulaire, et des interactions avec des enzymes ou des transporteurs. Aucun ne peut être tenu pour l’unique réponse, mais ensemble ils montrent pourquoi le mythe simpliste du récepteur échoue.
L’histoire pointe dans le même sens. Les travaux de Raphael Mechoulam sur le système endocannabinoïde ont ouvert un champ centré sur l’anandamide et le 2-AG, mais même l’anandamide n’est pas simplement un ligand de CB1. Elle active aussi TRPV1, le récepteur à la chaleur et à la capsaïcine dont l’importance sensorielle plus large a été reconnue par le prix Nobel de physiologie ou médecine 2021 décerné à David Julius et Ardem Patapoutian pour leurs découvertes sur les récepteurs de la température et du toucher. Dès qu’un cannabinoid endogène peut signaler à la fois via un GPCR et un canal TRP, le modèle « CB1/CB2 seulement » n’est plus un modèle. C’est une caricature.
Polypharmacologie : un ligand, de nombreuses cibles
Un meilleur point de départ est la polypharmacologie. Un seul ligand, de nombreuses cibles, avec des affinités, efficacités, tissus et conséquences différents. En pharmacologie, « sale » est parfois employé péjorativement, mais pour les cannabinoid, le terme est souvent simplement descriptif.
Considérons le nombre de types d’action rassemblés sous le même terme générique. THC est un agoniste partiel de CB1/CB2, mais des travaux de 2025 mis en avant par l’Université hébraïque ont souligné que THC inhibe les nocicepteurs périphériques en ciblant les canaux sodiques nociceptifs NaV1.7 et NaV1.8. Ce n’est plus de l’agonisme récepteur. C’est une inhibition de canal ionique sur des cibles déjà considérées comme des candidats majeurs pour des médicaments antalgiques. Si cette ligne de travaux se confirme à travers les espèces et les conditions de dosage, une partie de l’analgésie du THC pourrait venir d’un mécanisme qui ressemble davantage à un frein local de l’excitabilité qu’à un effet classique sur les récepteurs cannabinoid.
CBD présente une autre forme de promiscuité. Dans différents systèmes d’essais, on lui a attribué des effets sur TRPV1, TRPA1, TRPM8, 5-HT1A, PPAR-gamma, GPR55 et le tonus adénosinergique, entre autres. Le problème n’est pas un manque de mécanismes. Le problème est d’identifier lesquels comptent aux concentrations atteintes cliniquement chez l’humain. La reconnaissance de cibles in vitro est facile. Sa traduction est difficile. Un effet à l’échelle micromolaire dans une lignée cellulaire surexprimant un récepteur n’explique pas automatiquement les résultats chez le patient après administration orale, métabolisme de premier passage, liaison aux protéines et partitionnement tissulaire.
D’autres phytocannabinoid compliquent encore davantage le tableau. CBG a été décrit dans certains systèmes comme un composé agissant sur les récepteurs alpha-2 adrénergiques, les canaux TRP et 5-HT1A. CBC a été associé à TRPA1 et aux canaux TRPV. THCV peut se comporter différemment de delta-9-THC au niveau de CB1 selon la dose et le contexte, tout en conservant des possibilités non liées à CB1. Les cannabinoid acides tels que CBDA et THCA posent des questions supplémentaires, car la décarboxylation, la stabilité et la formation de métabolites modifient toutes l’exposition aux cibles. La même étiquette de flacon peut donc masquer des pharmacologies très différentes dès lors que la voie d’administration, la chaleur, le métabolisme et la formulation entrent en ligne de compte.[3]Library Docking for Cannabinoid-2 Receptor Ligands. American Chemical Society. Journal of Medicinal Chemistry, 2016. https://pubs.acs.org/doi/10.1021/acs.jmedchem.6c00835
Même dans la pharmacologie des GPCR, le domaine a dépassé les étiquettes grossières. GPR55 est encore parfois appelé candidat « CB3 », mais cela reste contesté pour de bonnes raisons ; le signal, l’ensemble des ligands et le rôle physiologique ne se superposent pas proprement aux récepteurs cannabinoid classiques. GPR18 et GPR119 sont également évoqués dans la littérature adjacente aux cannabinoid, notamment autour de l’inflammation, du métabolisme et de la signalisation intestinale, mais les preuves sont inégales. Les chimistes médicinaux le savent. Un article de 2016 du Journal of Medicinal Chemistry, « Library Docking for Cannabinoid-2 Receptor Ligands », illustrait une approche fondée sur la structure qui est presque l’opposé de la lore populaire sur les récepteurs : conception sélective des cibles, docking, optimisation des squelettes et séparation intentionnelle des effets souhaités des effets indésirables. Le domaine ne demande pas : « Est-ce que cela touche les récepteurs cannabinoid ? » Il demande : quelles cibles, dans quel état, dans quel tissu, à quelle concentration, et avec quel biais ?
Pourquoi les cibles autres que CB1/CB2 comptent cliniquement
C’est ici que la science cesse d’être sémantique et commence à influencer la médecine.
Pour la douleur, les cibles non CB1 peuvent être la voie la plus plausible vers des médicaments utiles avec moins d’intoxication. TRPV1, TRPA1, les canaux sodiques périphériques et les voies de transcription inflammatoire offrent tous des moyens de réduire le tir des nocicepteurs ou la sensibilisation neuro-immune sans forte activation centrale de CB1. Un article de ScienceDaily de 2026 sur un composé de cannabis qui « relieves pain without the high » ne constitue qu’un signal au stade de la recherche, pas une réponse clinique achevée, mais la direction est logique. Si l’analgésie peut être orientée vers des canaux ioniques périphériques ou une exposition tissulaire restreinte, l’ancien compromis entre soulagement de la douleur et charge psychoactive pourrait s’atténuer.
Pour l’inflammation et le métabolisme, PPAR-gamma est un bon exemple de l’importance des catégories de récepteurs. Les PPAR sont des récepteurs nucléaires, et non des récepteurs cannabinoid membranaires. Leur activation modifie des programmes d’expression génique liés au métabolisme des lipides, à la sensibilité à l’insuline et au tonus inflammatoire. Certains effets des cannabinoid rapportés dans des modèles métaboliques ou inflammatoires correspondent mieux à cette biologie transcriptionnelle lente qu’à une signalisation rapide via CB1. Mais là encore, la concentration et l’accès intracellulaire importent. Un article montrant l’activation de PPAR dans un essai rapporteur ne prouve pas un effet anti-inflammatoire cliniquement pertinent chez l’humain.[4]MIRA Pharmaceuticals Reports New Preclinical Data Demonstrating MIRA-55's Differentiated Mechanism of Action and Anxiolytic Activity Relative to THC. MIRA Pharmaceuticals. Nasdaq press release, 2025. https://www.nasdaq.com/press-release/mira-pharmaceuticals-reports-new-preclinical-data-demonstrating-mira-55s
Pour l’anxiété et les nausées, les mécanismes liés à la sérotonine réapparaissent sans cesse, en particulier 5-HT1A. Les données sont mixtes et souvent indirectes, mais la persistance du signal est révélatrice. Il est difficile de faire correspondre la réputation anxiolytique du CBD uniquement à CB1/CB2. C’est l’une des raisons pour lesquelles les entreprises tentent d’ingénier des composés inspirés des cannabinoid, différenciés, plutôt que de simplement fabriquer des analogues plus puissants du THC. En 2025, MIRA Pharmaceuticals a rapporté des données précliniques affirmant que son candidat MIRA-55 présentait un « differentiated mechanism of action » et une « anxiolytic activity relative to THC ». Les communiqués d’entreprise constituent un niveau de preuve faible et doivent être traités comme tel. Néanmoins, ils indiquent la direction du développement du médicament : s’éloigner de l’idée que la meilleure médecine cannabinoid n’est qu’une stimulation CB1 plus propre.
Le prurit, la migraine, l’épilepsie, les troubles intestinaux et la neuroprotection se situent tous dans la même zone mécanistique. Les canaux TRP régulent le gain sensoriel. Les GPCR orphelins peuvent façonner la signalisation immunitaire et épithéliale. Les PPAR modifient les programmes inflammatoires. Les canaux sodiques contrôlent directement l’excitabilité. Les voies sérotoninergiques influencent l’anxiété, les vomissements et les réponses au stress. Une fois ces systèmes placés à côté de CB1 et CB2 plutôt qu’en dessous, de nombreux effets réels des cannabinoid deviennent moins mystérieux et plus banalement pharmacologiques.
Le modèle simpliste survit parce qu’il est facile. Le meilleur modèle survit au contact des données.
Le système endocannabinoïde face au paysage plus large des cibles des cannabinoid
Les écrits populaires sur le cannabis traitent souvent la pharmacologie comme une histoire à deux récepteurs : CB1 explique les effets psychoactifs, CB2 les effets immunitaires, et tout le reste n’est qu’un détail. Cette représentation est trop étroite au regard des preuves. Elle ne rend pas compte de la raison pour laquelle le cannabidiol ne peut pas être expliqué proprement par CB1 ou CB2, de la raison pour laquelle certains cannabinoid déclenchent brûlure ou analgésie via les canaux TRP, de la raison pour laquelle des récepteurs nucléaires intracellulaires comme PPAR-γ réapparaissent sans cesse dans les études sur l’inflammation, ni de la raison pour laquelle même THC peut affecter des canaux sodiques pertinents pour la douleur en dehors de la signalisation cannabinoid classique. Si le domaine veut expliquer la douleur, l’anxiété, l’inflammation, le contrôle des crises ou les problèmes de sécurité des nouveaux intoxiquants, le réductionnisme récepteur doit disparaître.
Le moment réglementaire le montre clairement. En 2025, HHS a déclaré que « 7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety » en soutenant une action de classement contre des produits à 7-OH renforcé. Cette déclaration ne concernait pas le cannabis, mais elle capture la même leçon pharmacologique : dès que des fabricants passent de constituants végétaux familiers à des intoxicants renforcés, semi-synthétiques ou structurellement modifiés, les simples étiquettes de catégorie deviennent inutiles. « THC-like » vous dit beaucoup moins que le profil cible, la puissance, les métabolites, la distribution tissulaire et l’activité hors cible.
Les cibles canoniques : CB1, CB2, anandamide et 2-AG
Le système endocannabinoïde canonique reste important. CB1 et CB2 sont des récepteurs couplés aux protéines G, principalement couplés à Gi/o, identifiés à la fin du XXe siècle et cartographiés en détail par des chercheurs tels que Ken Mackie et Vincenzo Di Marzo. CB1 est fortement exprimé dans le système nerveux central, notamment dans le cortex, l’hippocampe, les noyaux gris centraux et le cervelet, ce qui explique pourquoi l’agonisme partiel du THC y est lié à l’intoxication, aux effets mnésiques, à l’altération du contrôle moteur et aux changements d’appétit. CB2 est enrichi dans les cellules immunitaires et les tissus périphériques, bien qu’il ne soit pas absent du cerveau. L’activation de l’un ou l’autre récepteur réduit généralement la formation d’AMPc, module les canaux ioniques et modifie la libération des neurotransmetteurs.
Les ligands endogènes sont l’anandamide et le 2-arachidonoylglycérol, généralement abrégé en anandamide et 2-AG. Le groupe de Raphael Mechoulam a été central dans cette histoire : l’anandamide a été identifiée en 1992, puis le 2-AG peu après. Elles ne sont pas stockées dans des vésicules synaptiques comme les neurotransmetteurs classiques. Elles sont synthétisées à la demande à partir de précurseurs lipidiques membranaires et agissent souvent de manière rétrograde, en se déplaçant des cellules postsynaptiques vers les terminaisons présynaptiques pour diminuer la libération des neurotransmetteurs. L’anandamide est principalement dégradée par FAAH ; le 2-AG principalement par MAGL. Ce cycle biochimique constitue l’ossature du système endocannabinoïde.
Mais l’ossature n’est pas tout le squelette. L’anandamide est aussi un agoniste de TRPV1. CBD a une faible affinité directe pour CB1 et CB2 comparée au THC, tout en ayant clairement des actions cliniquement significatives ; la solution orale de cannabidiol approuvée par la FDA est indiquée pour les crises associées au syndrome de Lennox-Gastaut, au syndrome de Dravet et au complexe de sclérose tubéreuse chez les patients âgés d’un an et plus. Cette utilisation approuvée rappelle que les effets cliniquement pertinents des cannabinoid n’ont pas besoin de s’aligner sur un fort agonisme de CB1.
Ce qui compte comme cible cannabinoid
Une définition pratique vaut mieux qu’une définition puriste. Une cible cannabinoid est tout site moléculaire sur lequel un phytocannabinoid, un endocannabinoïde, un métabolite ou un squelette inspiré des cannabinoid se lie ou module fonctionnellement la signalisation à des concentrations susceptibles d’avoir une importance dans les cellules, les tissus, les animaux ou l’humain. Selon ce critère, le paysage s’élargit rapidement.
Les canaux TRP sont les exemples non CB les plus familiers. TRPV1, TRPA1, TRPV2 et TRPM8 reviennent sans cesse dans les articles sur les cannabinoid. Ce n’est pas un détail secondaire. David Julius et Ardem Patapoutian ont partagé le prix Nobel de physiologie ou médecine 2021 « for their discoveries of receptors for temperature and touch », un rappel que les canaux ioniques qui gouvernent la chaleur, le froid, l’irritation et la mécanosensation sont directement situés dans les voies de la douleur. L’anandamide active TRPV1. CBD, CBG, CBC et les cannabinoid acides ont tous montré une activité sur les canaux TRP in vitro, souvent avec des effets dépendants de la concentration, parfois biphasés. Un cannabinoid qui active d’abord TRPV1 peut ensuite le désensibiliser, produisant le paradoxe d’une irritation initiale suivie d’une analgésie.
Les PPAR élargissent encore le cadre. PPAR-α et PPAR-γ sont des récepteurs nucléaires qui régulent la transcription liée au métabolisme lipidique et à l’inflammation. Certains cannabinoid et lipides liés au système endocannabinoïde agissent ici directement ou après accumulation intracellulaire et métabolisme. Il s’agit d’effets plus lents, de régulation génique, et non d’une signalisation à l’échelle de la milliseconde comme celle de CB1. Cela compte pour les affirmations concernant l’inflammation chronique, qui prennent souvent plus de sens via la signalisation nucléaire que via l’activité aiguë des récepteurs cannabinoid synaptiques.
Puis viennent les GPCR orphelins ou encore débattus, en particulier GPR55, GPR18 et GPR119. GPR55 a été proposé à plusieurs reprises comme candidat « CB3 », et l’étiquette reste prématurée. Le récepteur existe ; la classification est discutée. CBD est souvent décrit comme antagoniste de GPR55 ou modulateur négatif dans des systèmes expérimentaux, tandis que certains lipides endogènes et ligands synthétiques peuvent l’activer. GPR18 et GPR119 apparaissent dans l’inflammation, le métabolisme et la signalisation immunitaire, mais les preuves sont inégales et les effets d’espèce peuvent être substantiels.
Les récepteurs de la sérotonine, surtout 5-HT1A, doivent aussi figurer sur cette carte élargie. La littérature anxiolytique et antiémétique du CBD implique souvent 5-HT1A, bien que le débat sur l’agonisme direct versus la facilitation indirecte soit toujours ouvert. Cette distinction importe. Un composé qui se lie faiblement à un récepteur mais modifie de façon fiable le comportement d’un circuit par des mécanismes allostériques ou de réseau peut malgré tout avoir des effets significatifs in vivo. La même prudence s’applique aux programmes précliniques rapportés par les entreprises : en 2025, MIRA Pharmaceuticals a déclaré que son candidat MIRA-55 possédait un « differentiated mechanism of action » et montrait une activité anxiolytique par rapport au THC. Cela ne confirme pas un bénéfice clinique, mais cela montre où va la chimie médicinale — loin d’une simple imitation du THC et vers une pharmacologie cannabinoid façonnée par les cibles.[5]Psychoactive cannabinoid THC inhibits peripheral nociceptors by targeting NaV1.7 and NaV1.8 nociceptive sodium channels. Hebrew University of Jerusalem cannabinoids research portal. Research portal summary, 2025. https://cannabinoids.huji.ac.il/publications/psychoactive-cannabinoid-thc-inhibits-peripheral-nociceptors-targeting[6]A cannabis compound that relieves pain without the high. ScienceDaily. ScienceDaily, 2026. https://www.sciencedaily.com/releases/2026/06/260619033343.htm
Les canaux sodiques méritent aussi une place ici. Un rapport de l’Université hébraïque de 2025 a identifié l’inhibition de nocicepteurs périphériques par THC via les canaux sodiques nociceptifs NaV1.7 et NaV1.8. C’est une découverte importante parce que NaV1.7 et NaV1.8 sont des cibles centrales de la douleur, et que le mécanisme se situe en dehors de CB1/CB2. Cela s’inscrit aussi dans une dynamique translationnelle plus large. En 2026, ScienceDaily a mis en avant des travaux sur « a cannabis compound that relieves pain without the high ». Le composé exact et ses perspectives cliniques exigent un examen attentif, mais l’orientation est crédible : l’analgésie peut, en principe, être dissociée de l’intoxication centrale en ciblant des voies périphériques ou non CB1.
Biais de signalisation Propriété d’un ligand par laquelle il stabilise des états du récepteur qui favorisent une voie en aval plutôt qu’une autre, comme la signalisation par protéine G plutôt que le recrutement de bêta-arrestine.
Affinité, efficacité, biais et fenêtres de concentration
Cette carte des cibles plus large n’a de sens que si les termes pharmacologiques sont clairs. Ki est une constante d’affinité de liaison : un Ki plus faible signifie en général une liaison plus forte dans un essai de compétition. EC50 est la concentration produisant 50 pour cent de l’effet fonctionnel mesuré. Ce ne sont pas des équivalents. Un ligand peut se lier fortement tout en produisant une faible signalisation, ou se lier modérément tout en modifiant fortement la fonction par amplification de la voie.
Un agoniste active un récepteur. Un antagoniste bloque son activation par un autre ligand. Un agoniste inverse pousse des récepteurs constitutivement actifs vers une signalisation de base plus faible. THC au niveau de CB1 est généralement décrit comme un agoniste partiel : même lorsqu’il occupe les récepteurs, il ne produit pas l’effet maximal d’un agoniste à forte efficacité. Cela aide à expliquer pourquoi différents cannabinoid, et même différents ligands synthétiques de CB1, peuvent avoir des plafonds physiologiques très différents.
Le biais de signalisation signifie qu’un ligand stabilise des conformations du récepteur qui favorisent une voie plutôt qu’une autre, par exemple la signalisation par protéine G plutôt que le recrutement de β-arrestine. C’est désormais une pensée standard en développement de médicaments, y compris en chimie médicinale des cannabinoid ; l’article de 2016 du Journal of Medicinal Chemistry « Library Docking for Cannabinoid-2 Receptor Ligands » s’inscrit dans cette tradition orientée vers la cible. La désensibilisation signifie qu’une activation répétée ou prolongée peut réduire la réactivité, un problème majeur pour les canaux TRP et pour CB1 lui-même. Enfin, l’engagement de cible spécifique au tissu signifie que le même composé peut toucher différentes cibles dans le cerveau, l’intestin, la peau, les cellules immunitaires ou les nerfs périphériques selon la concentration, la voie d’administration, le métabolisme et l’expression locale des protéines. C’est pourquoi la promiscuité in vitro n’équivaut pas automatiquement à une pertinence clinique — mais c’est aussi pourquoi les explications limitées à CB1/CB2 échouent sans cesse.
Canaux TRP : les capteurs de chaleur, de douleur et d’irritation que les cannabinoid continuent d’atteindre
L’abrégé habituel dit que les cannabinoid agissent via CB1 et CB2. C’est trop étroit pour expliquer ce que font réellement beaucoup de ces molécules dans les tissus. À maintes reprises, les phytocannabinoid ciblent les canaux transitoires de potentiel récepteur, une superfamille de canaux ioniques présents dans les nocicepteurs, les kératinocytes, les nerfs des voies aériennes, les cellules immunitaires et d’autres interfaces sensorielles où l’organisme détecte la chaleur, le froid, les produits chimiques, l’étirement, les lésions et l’inflammation.[7]The Nobel Prize in Physiology or Medicine 2021. The Nobel Assembly at Karolinska Institutet. Nobel Prize Press Release, 2021. https://www.nobelprize.org/prizes/medicine/2021/press-release/
Cette biologie n’a rien d’obscur. Elle était suffisamment centrale pour la science somatosensorielle pour que le prix Nobel de physiologie ou médecine 2021 soit décerné à David Julius et Ardem Patapoutian « for their discoveries of receptors for temperature and touch ». Les travaux de Julius identifiant le récepteur de la capsaïcine, TRPV1, ont contribué à établir la vision moderne selon laquelle la signalisation de la douleur n’est pas simplement un fil transportant l’information de dommage ; elle est chimiquement régulée au tout premier terminus sensoriel. Cela compte pour les cannabinoid, car plusieurs cannabinoid végétaux majeurs interagissent avec la même machinerie moléculaire qui répond au piment, à l’huile de moutarde, à la chaleur nocive, aux agents refroidissants, aux conditions acides et aux lipides inflammatoires.
Le résultat est une pharmacologie qui paraît confuse si l’on attend un récepteur et un effet. Elle devient plus logique si l’on pense en termes de contrôle du gain sensoriel. De nombreux cannabinoid sont des ligands de faible à moyenne puissance sur les récepteurs cannabinoid et, en même temps, des modulateurs directs des canaux TRP. Certains les activent. Certains les inhibent. Certains font les deux selon la concentration, l’espèce, la variante d’épissage, l’environnement membranaire et le fait que l’essai mesure l’entrée de calcium, le courant, la libération de neuropeptides ou le comportement chez l’animal.
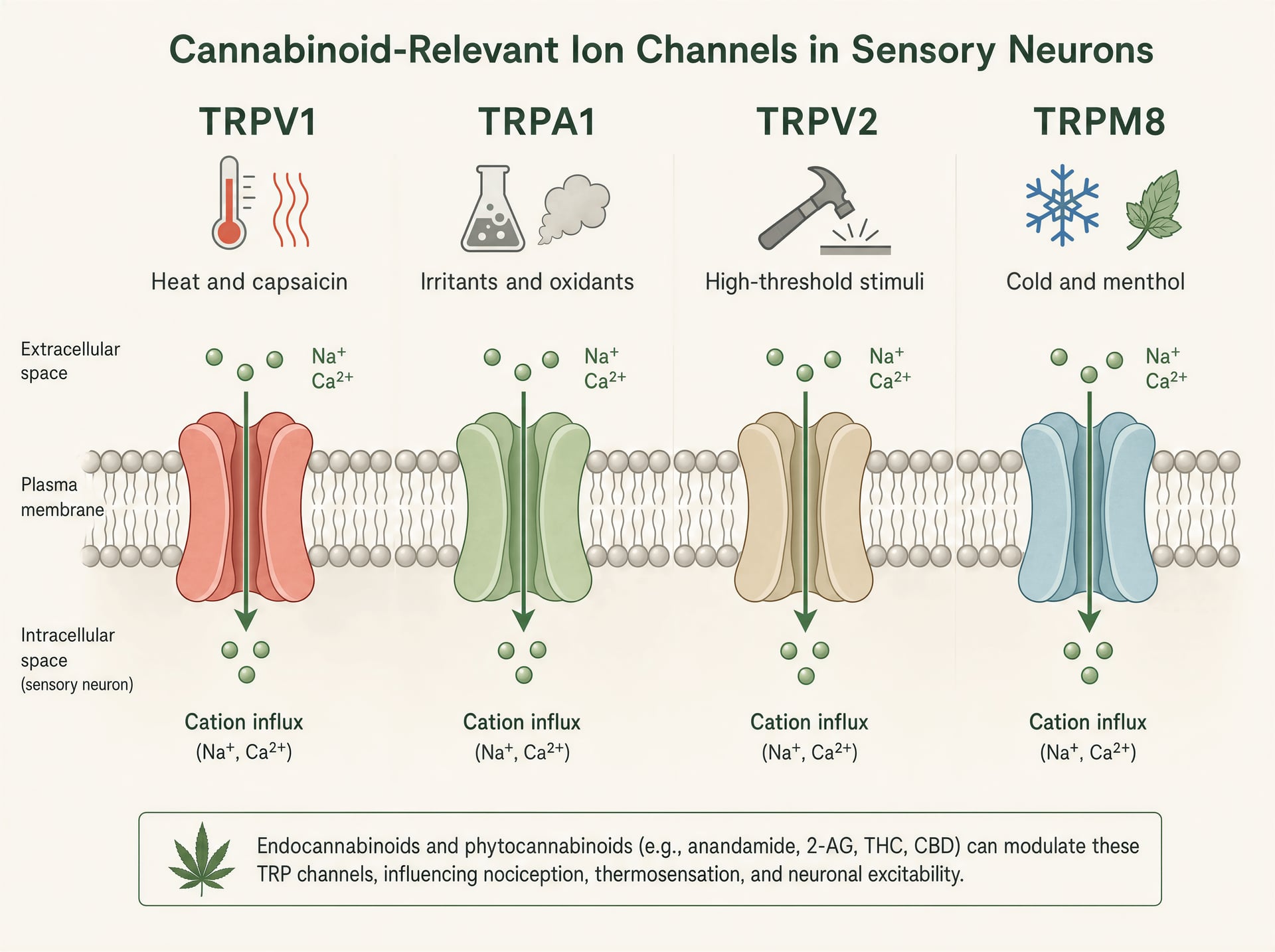
TRPV1, TRPA1, TRPV2 et TRPM8 dans la biologie sensorielle
Les canaux TRP sont des détecteurs polymodaux. TRPV1 est le mieux connu : activé par la capsaïcine, la chaleur nocive, les protons et les médiateurs inflammatoires endogènes, il est fortement exprimé dans les neurones sensoriels de petit diamètre qui génèrent la douleur brûlante et l’inflammation neurogène. Ouvrez le canal, les cations entrent, le neurone se dépolarise et le calcium intracellulaire augmente. TRPA1 se trouve souvent dans des populations nociceptives chevauchantes et est célèbre pour détecter des irritants électrophiles tels que l’isothiocyanate d’allyle provenant de la moutarde et du wasabi, l’acroléine de la fumée et les produits de stress oxydatif générés lors de l’inflammation. Il est pertinent non seulement pour la douleur, mais aussi pour le prurit, la toux, l’hyperréactivité des voies aériennes et la signalisation trigéminale de type migraineux.
TRPV2 est moins simple. C’est un canal thermo- et mécanosensible à haut seuil dans certains systèmes, mais il est aussi présent dans les cellules immunitaires, les glies et les tissus prolifératifs, ce qui explique qu’il revienne dans les discussions sur l’inflammation et, plus spéculativement, sur la biologie du cancer. TRPM8, à l’inverse, est le capteur canonique du froid, activé par les basses températures et des composés comme le menthol et l’iciline. Pourtant, il est aussi important dans les états douloureux, où l’allodynie au froid peut devenir sévère, et, dans certains contextes, l’activité de TRPM8 peut supprimer la douleur via une contre-stimulation circuitale. Même famille, rôles sensoriels très différents.
Cet éventail de fonctions explique pourquoi les effets des cannabinoid peuvent sembler contradictoires en surface. Activer TRPV1 ou TRPA1 peut piquer. Bloquer TRPM8 peut réduire les sensations de refroidissement mais aussi modifier la douleur au froid. Stimuler TRPV2 dans un type cellulaire peut affecter la signalisation calcique sans produire aucun effet sensoriel évident. Il n’existe pas un seul « effet TRP » de plus qu’il n’existe un seul « effet cannabinoid ».
CBD, CBG, CBC et THC sur les canaux de la famille TRP
Parmi les phytocannabinoid, CBD présente le profil TRP le plus solide et le mieux répliqué. Dans des systèmes d’expression hétérologues, CBD active TRPV1, TRPA1 et TRPV2 humaines à des concentrations micromolaires et inhibe TRPM8. Une étude largement citée de De Petrocellis et collègues en 2011, utilisant l’imagerie calcique dans des cellules HEK-293 transfectées, a montré que CBD se comportait comme agoniste de TRPV1, TRPV2, TRPA1 et TRPV4, tout en antagonisant TRPM8. La puissance n’était pas uniforme : TRPA1 était particulièrement sensible, avec une activité dans la faible gamme micromolaire, alors que d’autres canaux exigeaient des concentrations un peu plus élevées. Ce schéma a suffisamment résisté pour que l’implication des TRP fasse désormais partie de toute analyse sérieuse de la pharmacologie de CBD.
CBG et CBC s’inscrivent dans la même tendance générale, avec leur propre signature. CBG a montré à plusieurs reprises une activité sur TRPA1 et TRPV1, ainsi qu’une inhibition de TRPM8, ce qui le rend pharmacologiquement intéressant pour les modèles de douleur inflammatoire et d’hypersensibilité viscérale. CBC est moins étudié que CBD, mais les travaux in vitro disponibles suggèrent qu’il active lui aussi TRPA1 et peut engager TRPV1. Il ne s’agit pas de petites curiosités observées dans un seul essai puis jamais revues. Elles reviennent dans des systèmes recombinants et des préparations sensorielles primaires, précisément pour cette raison elles réapparaissent sans cesse dans les articles mécanistiques sur l’analgésie et l’inflammation.
THC est plus compliqué. Il peut activer TRPV2 et a été rapporté comme interagissant avec TRPA1 et TRPV1 dans certaines conditions, mais sa pharmacologie est dominée dans de nombreuses expériences par des effets médiés par CB1, surtout dans le système nerveux central. Néanmoins, l’idée que THC n’est qu’un médicament CB1 est fausse. Des travaux récents de l’Université hébraïque, rapportés en 2025, ont soutenu que THC inhibe les nocicepteurs périphériques en ciblant les canaux sodiques NaV1.7 et NaV1.8, un mécanisme non CB distinct qui cadre avec le point général développé ici : les cannabinoid touchent souvent plusieurs cibles liées à la douleur à la fois. Les canaux TRP font partie de cette carte élargie hors CB.
Une mise en garde s’impose. Une grande partie de ces preuves provient d’essais micromolaires, et toutes les concentrations micromolaires en boîte ne correspondent pas à une concentration libre atteignable sur une cible humaine. Les cannabinoid lipophiles se partitionnent dans les membranes, se lient aux protéines et génèrent des métabolites ; la voie d’administration et l’accumulation tissulaire comptent. Le fait que la solution orale de CBD soit approuvée par la FDA pour des syndromes épileptiques ne prouve pas que TRPV1 ou TRPA1 soient à l’origine de ses effets cliniques dans l’épilepsie. Cela montre simplement que CBD produit clairement chez l’humain des effets qu’on ne peut pas saisir en l’appelant un « composé agoniste des récepteurs cannabinoid non intoxicant ». Le récit moléculaire est plus vaste que cette étiquette.
L’activité TRP est aussi sensible à l’essai. Un canal peut sembler « activé » dans un test calcique parce que les réserves intracellulaires, le potentiel membranaire ou les lipides endogènes changent en parallèle. Les différences d’espèce peuvent être réelles. L’état fonctionnel aussi. Les tissus inflammés s’acidifient, s’oxydent et produisent des médiateurs lipidiques, qui modifient tous le gating des TRP. Un cannabinoid qui modifie à peine un canal à l’état basal peut avoir un effet beaucoup plus important dans une terminaison nerveuse lésée.
Désensibilisation, analgésie et pourquoi l’activation peut réduire la douleur
C’est l’élément qui déroute les non-spécialistes : si TRPV1 et TRPA1 sont des canaux générateurs de douleur, pourquoi leur activation réduirait-elle jamais la douleur ?
Parce que l’activation aiguë et la sortie fonctionnelle soutenue ne sont pas identiques.
TRPV1 est l’exemple classique. La capsaïcine brûle d’abord, puis désensibilise les nocicepteurs et peut produire une analgésie après une exposition répétée ou à forte concentration. Cliniquement, ce principe est utilisé dans le patch de capsaïcine à 8 pour cent pour la douleur neuropathique. Le mécanisme inclut une désensibilisation dépendante du calcium, l’épuisement de neuropeptides tels que substance P et CGRP, une modification de l’état de phosphorylation du canal et, dans certains cas, une défonctionnalisation réversible de la terminaison nerveuse. Un canal qui s’active fortement au début peut devenir moins réactif ensuite. Le signal immédiat est pronociceptif ; l’état subséquent peut être antinociceptif.
Les cannabinoid semblent exploiter cette même logique. L’activation de TRPV1 ou TRPA1 par CBD peut déclencher une entrée de calcium, suivie d’une diminution de la réactivité du canal et d’un affaiblissement de l’excitabilité des neurones sensoriels. C’est une voie plausible par laquelle un composé peut piquer dans une boîte de Pétri tout en réduisant l’hyperalgésie chez l’animal. L’axe temporel compte. La dose aussi. De faibles concentrations peuvent sensibiliser ou activer faiblement. Des concentrations plus élevées peuvent induire une désensibilisation ou même des effets membranaires plus larges qui suppriment le tir neuronal.
TRPA1 ajoute une couche supplémentaire, car il est profondément lié aux irritants inflammatoires et au stress oxydatif. Dans les systèmes des voies aériennes et trigéminaux, une activation répétée ou prolongée peut modifier la libération des neuropeptides et la réactivité réflexe. Cela le rend pertinent pour la toux, la migraine et les poussées inflammatoires, pas seulement pour la « douleur » au sens étroit. Si un cannabinoid engage TRPA1 puis diminue la réactivité ultérieure, l’effet net peut être une réduction de la signalisation irritative, même si le premier événement moléculaire a été l’ouverture du canal.
TRPM8 montre, dans de nombreux essais, le type d’effet cannabinoid inverse : des cannabinoid comme CBD et CBG l’inhibent souvent plutôt que de l’activer. Cela peut compter dans l’hypersensibilité au froid, où une signalisation excessive via TRPM8 contribue à une allodynie douloureuse au froid. Ici, il n’y a pas de paradoxe activation→soulagement ; l’hypothèse la plus simple est une suppression directe d’une voie de détection du froid. Mais il ne faut pas non plus surestimer cela. Dans certains états douloureux, l’activité de TRPM8 peut contrebalancer la douleur à la chaleur ou le prurit, de sorte que son blocage n’est pas automatiquement bénéfique.
La position la plus solide que les preuves permettent est la suivante : les canaux TRP ne sont pas des notes de bas de page dans la pharmacologie des cannabinoid. Ce sont des cibles récurrentes, fonctionnellement pertinentes, surtout pour les effets sensoriels périphériques impliquant la chaleur, l’irritation chimique, la douleur inflammatoire, le prurit et les réflexes des voies aériennes. Ils n’expliquent pas tout. Ils ne sont pas toujours le mécanisme dominant in vivo. Néanmoins, quiconque tente de comprendre pourquoi CBD, CBG, CBC, ou même THC peut modifier la douleur et l’inflammation sans correspondance nette avec CB1 ou CB2 doit voir TRPV1, TRPA1, TRPV2 et TRPM8 apparaître tôt dans le raisonnement, et non comme une réflexion tardive.
Cela compte aussi pour le développement de médicaments. Les agences de santé publique distinguent déjà les cannabinoid familiers des intoxicants chimiquement modifiés ou renforcés, car des différences au niveau des cibles peuvent modifier le risque. Le même principe vaut à l’inverse pour les thérapeutiques : si l’analgésie peut être séparée de l’intoxication centrale, une voie consiste à concevoir des composés qui orientent l’action vers les canaux TRP périphériques et d’autres cibles non CB plutôt que vers un agonisme CB1 fortement pénétrant dans le cerveau. L’ancien récit réductionniste des récepteurs est trop étroit pour les données.
PPAR : les cannabinoid comme signaux lipidiques intracellulaires, et non comme de simples ligands de récepteurs membranaires
Les récepteurs activés par les proliférateurs de peroxysomes, généralement abrégés en PPAR, changent la conversation sur les cannabinoid parce qu’ils se situent dans un autre endroit et fonctionnent sur un autre tempo que CB1 et CB2. CB1 et CB2 sont des récepteurs membranaires couplés aux protéines G, conçus pour une signalisation rapide : secondes à minutes, canaux ioniques, libération de neurotransmetteurs, cascades de kinases. Les PPAR sont des récepteurs nucléaires. Ils répondent à des molécules lipophiles, mobilisent l’appareil transcriptionnel et modifient les gènes qu’une cellule exprime sur des heures à des jours. Ce changement est important. Il signifie que certains effets des cannabinoid peuvent ressembler moins à un agonisme classique de récepteur et davantage à une reprogrammation régulée par les lipides du tonus inflammatoire, du traitement mitochondrial, de l’oxydation des acides gras, de la signalisation fibrotique et des réponses gliales.
Il ne s’agit pas d’une spéculation éloignée. Les cannabinoid sont très lipophiles, s’accumulent dans les membranes, se répartissent dans des compartiments intracellulaires et génèrent des métabolites qui peuvent avoir des profils de cibles différents de ceux de la molécule mère. Une classe de médicaments possédant ces propriétés est presque faite pour rencontrer des capteurs lipidiques nucléaires. Les PPAR figurent parmi les sites les plus plausibles où cela se produit.
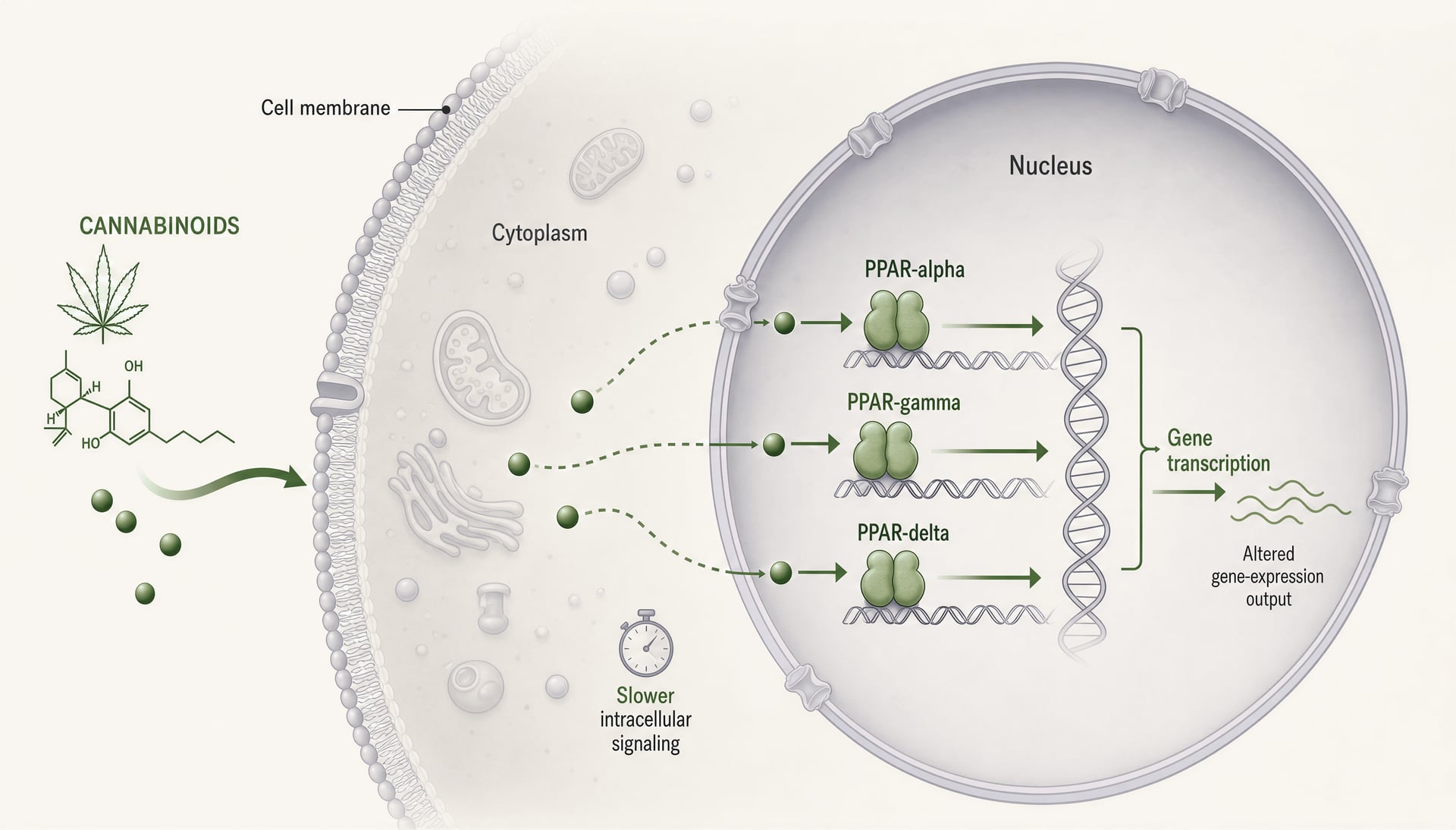
Ce que font PPAR-alpha, PPAR-gamma et PPAR-delta
Les trois principaux isoformes de PPAR se recoupent, mais ils ne sont pas interchangeables. PPAR-alpha est classiquement lié au catabolisme des acides gras. Il est abondant dans le foie, le cœur, le rein, le muscle et d’autres tissus qui brûlent les graisses de manière intensive, et lorsqu’il est activé, il oriente des programmes transcriptionnels vers la beta-oxydation, la cétogenèse, la gestion des lipoprotéines et une diminution de la signalisation inflammatoire. Les pharmacologues le connaissent à travers les fibrates. Dans la recherche sur la douleur et l’inflammation, PPAR-alpha compte aussi au-delà du métabolisme parce qu’il peut supprimer l’expression de gènes inflammatoires liés à NF-kappaB et modifier la signalisation sensorielle.
PPAR-gamma est l’isoforme qui revient sans cesse dans les articles sur les cannabinoid, parfois pour de bonnes raisons, parfois parce que c’est l’histoire la plus facile à raconter. Il est hautement pertinent pour la différenciation des adipocytes et la sensibilité à l’insuline, mais cette formule est réductrice. PPAR-gamma régule la polarisation des macrophages, la production de cytokines, les réponses au stress oxydatif, le remodelage fibrotique, le comportement endothélial et l’activation gliale dans le système nerveux central. Cela lui donne une pertinence évidente pour les maladies inflammatoires de l’intestin, la neuroinflammation, les complications du diabète et la fibrose tissulaire. C’est aussi une cible à double tranchant : une activation puissante peut améliorer la sensibilité à l’insuline tout en entraînant un œdème, une prise de poids et d’autres inconvénients familiers des thiazolidinediones.
PPAR-delta, aussi appelé PPAR-beta/delta, reçoit moins d’attention dans les textes grand public sur le cannabis, mais il ne devrait pas. Il est largement exprimé et soutient l’utilisation des acides gras, la fonction mitochondriale, la réparation des plaies, la biologie des kératinocytes et certains programmes anti-inflammatoires. Selon le contexte, il peut freiner ou faciliter des processus pathologiques, ce qui contribue à la moindre netteté de la littérature qui le concerne. Si un cannabinoid ou un métabolite de cannabinoid engage PPAR-delta, la lecture biologique peut varier beaucoup plus selon le tissu que ne le suggère une histoire simpliste « agoniste=bénéfice ».
Mécaniquement, les trois isoformes agissent comme des facteurs de transcription activés par un ligand, qui hétérodimérisent avec le récepteur X des rétinoïdes et se lient à des éléments de réponse aux proliférateurs de peroxysomes dans l’ADN. Une fois engagés, ils ne font pas qu’enclencher un interrupteur. Ils modifient des réseaux transcriptionnels. Les coactivateurs, les corépresseurs, l’état de la chromatine, le type cellulaire, le contexte inflammatoire et la conformation du récepteur propre au ligand influencent tous le résultat. Deux composés peuvent tous deux être appelés agonistes de PPAR-gamma tout en produisant une biologie sensiblement différente.
Ce point est particulièrement important pour les cannabinoid, qui sont souvent des molécules pharmacologiquement promiscues plutôt que des outils propres et à cible unique.
CBD et cannabinoid apparentés dans la signalisation métabolique et inflammatoire
CBD est l’exemple récurrent parce que son profil clinique s’explique mal par CB1 ou CB2 seuls. La solution orale de CBD approuvée par la FDA pour les crises dans le syndrome de Lennox-Gastaut, le syndrome de Dravet et le complexe de sclérose tubéreuse montre que CBD est pharmacologiquement réel chez l’humain, mais pas qu’une seule cible non cannabinoid explique ses actions. PPAR-gamma est l’un des candidats les plus cités parce que plusieurs études cellulaires et animales ont relié CBD à des effets anti-inflammatoires et métaboliques atténués par des antagonistes de PPAR-gamma ou accompagnés de modifications transcriptionnelles dépendantes de PPAR-gamma.
Un article très cité d’O’Sullivan et collègues en 2009 a rapporté que CBD provoquait une vasorelaxation dans des artères humaines et qu’une partie de l’effet était sensible à l’antagoniste de PPAR-gamma GW9662, suggérant une composante dépendante de PPAR-gamma. En 2011, Esposito et co-auteurs ont montré dans un modèle cellulaire de type Alzheimer que CBD réduisait la neuroinflammation induite par le beta-amyloïde et que le blocage de PPAR-gamma diminuait cet effet protecteur. En 2013, Hind et O’Sullivan ont passé en revue les preuves indiquant que les cannabinoid peuvent activer les PPAR directement ou indirectement, en intégrant CBD, THC, l’acide ajulémique, des lipides apparentés à l’anandamide et plusieurs cannabinoid synthétiques.
Le schéma est assez cohérent pour être pris au sérieux : CBD se retrouve souvent dans des systèmes expérimentaux où les gènes inflammatoires diminuent, les marqueurs de stress oxydatif baissent et l’antagonisme de PPAR-gamma affaiblit la réponse. Mais le prendre au sérieux ne revient pas à le tenir pour établi. Beaucoup de ces études utilisent des concentrations micromolaires de CBD. Cela compte parce que les concentrations libres intracellulaires dans les tissus humains vivants sont difficiles à déduire des concentrations nominales dans une boîte. CBD se lie aussi aux membranes et les perturbe, influence la gestion du calcium, interagit avec les canaux TRP, affecte la signalisation de l’adénosine en inhibant le transport des nucléosides et peut modifier le tonus endocannabinoïde. Chacune de ces voies peut alimenter des modifications transcriptionnelles qui ressemblent ensuite à un effet « de type PPAR ».
Les cannabinoid apparentés renforcent le dossier sans le rendre plus simple. THC a été rapporté dans certains systèmes comme activateur de PPAR-gamma, bien que généralement faiblement comparé aux ligands dédiés. L’acide cannabidiolique et l’acide tétrahydrocannabinolique ont montré une activité sur PPAR dans certains essais. Les lipides apparentés au système endocannabinoïde, tels que le palmitoyléthanolamide, l’oléoyléthanolamide et certains dérivés oxydés, entretiennent des relations plus fortes et mieux établies avec PPAR-alpha et PPAR-gamma que les phytocannabinoid les plus connus. C’est l’une des raisons pour lesquelles le cadre de la signalisation lipidique intracellulaire est meilleur qu’un cadre étroit du type « les cannabinoid végétaux se lient aux PPAR ». L’espèce active peut être le cannabinoid parent, un métabolite, un médiateur lipidique coadministré, ou une modification en aval des pools lipidiques endogènes.
L’acide ajulémique constitue une étude de cas utile. C’est un analogue synthétique apparenté au THC, développé intentionnellement pour s’éloigner de l’intoxication classique. Dans différents travaux précliniques, il a montré des actions anti-inflammatoires et antifibrotiques, avec des éléments impliquant PPAR-gamma parmi d’autres cibles. Ce type de chimie médicinale reflète une tendance plus large du domaine. Dès 2016, un article de l’ACS Journal of Medicinal Chemistry intitulé « Library Docking for Cannabinoid-2 Receptor Ligands » reflétait déjà une ingénierie ciblée fondée sur la structure plutôt qu’une simple étiquette récepteur, et les programmes cannabinoid plus récents visent de plus en plus à séparer l’analgésie, l’anxiolyse ou l’immunomodulation de l’activation centrale de CB1. La même logique s’applique aux squelettes actifs sur les PPAR : si une biologie cannabinoid utile peut être extraite par des mécanismes transcriptionnels et périphériques, aucune raison ne veut que le développement de médicaments reste prisonnier de la pharmacologie de type THC.
Les données de CBD sur la signalisation métabolique sont plus mitigées que ses données anti-inflammatoires. Certaines études précliniques suggèrent une meilleure sensibilité à l’insuline, une réduction des adipokines inflammatoires ou une meilleure gestion mitochondriale. D’autres ne montrent pas de bénéfice majeur, et les preuves humaines sont faibles. Le discours public va souvent plus vite que les données à ce sujet. Le fait que PPAR-gamma contrôle le métabolisme du glucose et du tissu adipeux ne signifie pas que CBD soit un modulateur métabolique cliniquement significatif chez l’humain aux expositions habituelles.
Transcription génique, effets retardés et limites des preuves
La biologie des PPAR impose une correction de l’échelle temporelle. Si un effet cannabinoid apparaît en quelques secondes ou quelques minutes, les PPAR ne sont probablement pas l’explication principale. La signalisation par récepteur nucléaire exige généralement un accès du ligand à des compartiments intracellulaires, l’engagement du récepteur, une modification du recrutement des co-régulateurs, des changements transcriptionnels, puis des conséquences au niveau protéique. Cela prend du temps. Des heures sont plausibles. Des jours sont courants. Lorsque des articles affirment qu’un effet rapide du cannabinoid est « via PPAR-gamma », le scepticisme est de mise à moins que la conception de l’étude ne sépare clairement la signalisation non génomique immédiate des effets transcriptionnels ultérieurs.
La conception des essais est le problème récurrent. Les essais rapporteurs peuvent montrer qu’un composé augmente la transcription dépendante des PPAR, mais les systèmes rapporteurs sont artificiels et peuvent exagérer une faible activité. Les études utilisant des antagonistes sont utiles, mais des médicaments comme GW9662 ne constituent pas un sérum de vérité ; les effets hors cible et le blocage partiel compliquent l’interprétation. Les essais de liaison aident, mais une liaison directe ne garantit pas que l’exposition tissulaire atteigne la concentration requise in vivo. Les modèles knock-out sont plus solides, même si la compensation par d’autres voies peut brouiller les résultats. La meilleure preuve combine les méthodes : engagement direct de la cible, pharmacologie sélective du récepteur, perturbation génétique, concentrations tissulaires pertinentes et cinétique compatible avec une action transcriptionnelle. Une grande partie de la littérature cannabinoid-PPAR n’atteint pas ce niveau.
La place centrale de PPAR-gamma dans la recherche sur CBD est donc à la fois justifiée et exagérée. Justifiée, parce que le signal se répète dans des modèles vasculaires, inflammatoires, neurodégénératifs et fibrosants. Exagérée, parce que CBD est précisément le type de molécule lipophile, à cibles multiples, pour lequel la concentration intracellulaire, les métabolites actifs et le contexte expérimental peuvent produire des récits mécanistiques séduisants mais incomplets. Une baisse du TNF-alpha ou de l’IL-6 après exposition à CBD n’est pas une empreinte digitale. C’est un indice.
Il n’en reste pas moins que le point plus large tient. Les cannabinoid ne doivent pas être considérés uniquement comme des ligands des récepteurs cannabinoid membranaires. Certains agissent, directement ou indirectement, comme signaux lipidiques intracellulaires qui peuvent engager la machinerie transcriptionnelle nucléaire. Cela ouvre des voies plausibles vers des effets anti-inflammatoires, antifibrotiques et neuro-immunitaires plus lents, moins liés à l’intoxication, et potentiellement plus pertinents pour la modification à long terme des maladies que la signalisation aiguë de CB1. Cela impose aussi une leçon réglementaire. Comme les autorités l’ont souligné dans d’autres contextes, y compris la déclaration du HHS en 2025 selon laquelle les produits à 7-hydroxymitragynine renforcé représentent « an imminent hazard to public safety », les différences au niveau moléculaire comptent. De petits changements structuraux peuvent rediriger l’engagement des cibles. Pour les cannabinoid et les produits apparentés, cela signifie que l’histoire de la sécurité et de l’efficacité ne peut pas être déduite de la seule familiarité avec THC, et la biologie des PPAR est l’une des raisons.
GPR55, GPR18 et GPR119 : le problème des GPCR orphelins
Un GPCR orphelin est un récepteur couplé aux protéines G dont le ligand endogène, le rôle physiologique, ou les deux restent incertains. Un récepteur « déorphanisé » est un récepteur pour lequel un activateur endogène convaincant a été proposé et répliqué au point de soutenir une biologie de travail. Cela semble ordonné. En pratique, ce l’est rarement. La pharmacologie des cannabinoid se heurte sans cesse à ce désordre parce que les endocannabinoid et les phytocannabinoid sont lipophiles, actifs sur les membranes et promiscues : ils peuvent modifier le flux calcique, l’activité des kinases ou la transcription de manière qui paraît médiée par un récepteur alors que la cible directe reste incertaine. C’est ainsi que GPR55, GPR18 et GPR119 sont entrés dans la conversation comme « récepteurs cannabinoid non classiques ».
La tentation de créer un nouveau label de récepteur est forte. Cela fait les titres. Cela dépasse aussi les preuves. GPR55 est celui qui s’est le plus rapproché du statut de « CB3 », mais le domaine n’a jamais atteint la cohérence qui soutient CB1 et CB2. La même prudence vaut encore davantage pour GPR18 et GPR119.
Pourquoi GPR55 a autrefois été appelé un possible récepteur cannabinoid
GPR55 a été cloné en 1999, et les premières études d’expression l’ont placé dans des tissus pertinents pour la biologie cannabinoid : régions cérébrales, ganglions rachidiens dorsaux, rate, tractus gastro-intestinal, système vasculaire, cellules immunitaires et cellules osseuses, y compris des ostéoclastes et des populations de la lignée ostéoblastique. Cette distribution comptait. Un récepteur exprimé dans les voies de la douleur, les tissus inflammatoires et l’os invite immédiatement à la comparaison avec CB1 et CB2, surtout lorsque des ligands cannabinoid semblent modifier ses lectures.
Son profil de signalisation paraissait aussi suffisamment différent pour être intéressant. Contrairement à CB1 et CB2, qui sont principalement couplés à Gi/o et inhibent généralement l’adénylyl cyclase, GPR55 signale le plus souvent via Gα12/13 et parfois via des voies liées à Gq, activant RhoA, la phospholipase C, ERK et la libération calcique intracellulaire. Dans les essais cellulaires, la lecture caractéristique est souvent un transitoire calcique. Cela rendait GPR55 facile à « voir » dans des systèmes hétérologues, mais aussi facile à surinterpréter, car les essais calciques sont sensibles à la densité du récepteur, au contexte cellulaire, à la lipophilie du ligand et au timing de l’essai.
La raison spécifique pour laquelle GPR55 est devenu un candidat récepteur cannabinoid était que plusieurs cannabinoid et ligands apparentés produisaient des effets mesurables à son niveau. Ryberg et collègues, dans le British Journal of Pharmacology en 2007, ont rapporté que GPR55 pouvait être activé par plusieurs ligands cannabinoid et l’ont proposé comme « a novel cannabinoid receptor ». Cet article est devenu la charnière historique. Il n’a pas réglé la question ; il l’a créée.
Peu après, les fissures sont apparues. Certains groupes ont trouvé que le lysophosphatidylinositol, en particulier les espèces 2-arachidonoyl LPI, constituait un agoniste endogène plus convaincant que n’importe quel cannabinoid classique. Oka et collègues en 2007, puis des travaux ultérieurs, ont fortement défendu cette idée. D’autres ont observé que les composés souvent discutés dans la recherche sur les cannabinoid se comportaient de manière incohérente sur GPR55 : le cannabidiol (CBD) apparaissait souvent comme antagoniste ou modulateur négatif dans certains essais, tandis que le Δ9-THC était faible, partiel ou inactif selon le système. L’« abnormal cannabidiol », O-1602 et certains cannabinoid synthétiques montraient parfois une activité plus nette que le THC lui-même. Ce n’est pas ce que l’on attend d’un troisième récepteur cannabinoid propre.
Néanmoins, la biologie de GPR55 est réelle, même si l’étiquette est instable. Dans la recherche sur la douleur, le récepteur est exprimé dans les neurones sensoriels et les circuits spinaux, et l’interruption génétique ou pharmacologique de la signalisation GPR55 a réduit l’hypersensibilité mécanique dans certains modèles murins. Staton et collègues dans Pain (2008) ont relié l’activation de GPR55 au traitement de la douleur inflammatoire et neuropathique, l’antagonisme réduisant l’hypersensibilité. Pourtant, l’effet n’est pas universel selon les modèles ou les ligands. Certaines données suggèrent une signalisation pronociceptive via la mobilisation du calcium et l’augmentation de l’excitabilité neuronale ; d’autres jeux de données sont plus faibles ou limités à certains modèles. La lecture la plus sûre est que GPR55 peut contribuer à la signalisation douloureuse dans certains contextes, en particulier inflammatoires, mais qu’il ne s’agit pas d’un interrupteur maître de la douleur.
La biologie osseuse fournit un signal plus solide. Pourquoi ? Parce que les phénotypes des souris knock-out pour GPR55 sont plus difficiles à attribuer à des artefacts d’essai. En 2009, Whyte et collègues ont rapporté dans PNAS que les souris dépourvues de GPR55 présentaient une masse osseuse accrue et une fonction ostéoclastique altérée, soutenant l’idée que GPR55 favorise la résorption ostéoclastique. Cela faisait sens compte tenu de sa signalisation liée au calcium et à RhoA. Les ostéoclastes dépendent de réarrangements cytosquelettiques et d’une gestion locale du calcium ; GPR55 correspond mieux à cette machinerie que CB1. Si un cannabinoid ou un composé apparenté module GPR55 ici, la conséquence physiologique pourrait être importante.
L’inflammation est le troisième grand thème. GPR55 est présent dans des cellules liées à l’immunité, et son activation a été associée à la libération de cytokines, au comportement des leucocytes et aux réponses inflammatoires vasculaires. Mais là encore, la direction n’est pas parfaitement uniforme. Dans certaines préparations, l’activation de GPR55 paraît pro-inflammatoire ; dans d’autres, plus régulatrice, ce qui reflète probablement le type cellulaire, le biais du ligand et les interactions croisées entre récepteurs plutôt qu’une contradiction simple. Un récepteur couplé à plusieurs voies et situé dans différents environnements membranaires ne produira pas une réponse universelle.
Cette complexité explique la longue lutte agoniste/antagoniste dans la littérature cannabinoid. CBD en est l’exemple le plus clair. Dans plusieurs études, CBD a souvent agi comme antagoniste ou inhibiteur fonctionnel de GPR55, atténuant la signalisation calcique induite par le LPI. Lauckner et collègues en 2008, dans un article très cité de PNAS, ont montré que l’activation de GPR55 augmentait le calcium intracellulaire et favorisait la libération des neurotransmetteurs, tandis que CBD contrecarrait certaines de ces voies de signalisation. Cela a alimenté une hypothèse durable selon laquelle certains effets du CBD, en particulier dans les modèles d’épilepsie et d’inflammation, pourraient impliquer en partie un blocage de GPR55 plutôt qu’une action sur CB1 ou CB2. Cette idée est plausible. Elle n’est pas démontrée comme mécanisme dominant chez l’humain.
THC est encore plus confus. Certains rapports le classent comme agoniste de faible puissance sur GPR55 ; d’autres ne trouvent qu’une efficacité négligeable ; d’autres encore suggèrent un comportement dépendant de la réserve réceptrice ou de la voie mesurée. Un ligand peut ressembler à un agoniste dans un essai β-arrestine, être neutre en liaison et antagoniste dans un essai calcique si le système est surexprimé ou biaisé. Ce n’est pas une note technique accessoire. C’est l’histoire même du problème.
Les preuves mixtes pour GPR18 et GPR119
GPR18 a souvent été discuté parce qu’il répond dans certains systèmes à N-arachidonoylglycine, un lipide lié aux endocannabinoid, et parce que l’« abnormal cannabidiol » et des composés apparentés ont montré des effets vasculaires ou immunitaires que certains auteurs ont rattachés à GPR18. Son expression a été rapportée dans les cellules immunitaires, la microglie, la rate et certains tissus périphériques. Cela le rendait attrayant comme candidat à la régulation de l’inflammation, de la migration immunitaire et éventuellement de la douleur.
Mais la pharmacologie a été inégale dès le départ. Kohno et collègues en 2006 ont soutenu l’activation de GPR18 par N-arachidonoylglycine. McHugh et collègues ont ensuite relié GPR18 à la migration microgliale et à la signalisation inflammatoire. Puis les problèmes de réplication sont apparus. Certains laboratoires ne parvenaient pas à reproduire les réponses aux ligands dans les systèmes transfectés. D’autres trouvaient une forte dépendance au marquage du récepteur, à la lignée cellulaire ou à l’orthologue d’espèce. Un récepteur qui « fonctionne » seulement dans une architecture d’essai n’est pas déorphanisé au sens stable du terme. Pour les cannabinoid spécifiquement, les preuves sont plus faibles que ne le suggèrent les résumés populaires. Il peut exister une vraie biologie ici, mais l’argument en faveur de GPR18 comme récepteur cannabinoid à part entière reste mince.
GPR119 est différent. Il est beaucoup moins plausible comme récepteur cannabinoid, malgré son inclusion occasionnelle dans des listes larges de récepteurs « non CB ». GPR119 est principalement associé à la détection des lipides dans les cellules bêta pancréatiques et les cellules entéroendocrines, couplé à Gs pour augmenter l’AMPc et favoriser la sécrétion d’insuline dépendante du glucose et la libération d’incrétines. L’oléoyléthanolamide est un candidat ligand endogène mieux établi que n’importe quel cannabinoid classique. Parce que certains éthanolamides d’acides gras sont structurellement proches de la chimie endocannabinoïde, GPR119 peut être entraîné dans les discussions sur le cannabis par association. C’est surtout une confusion de catégories. Le chevauchement est une proximité chimique, pas une preuve forte que THC, CBD ou les principaux phytocannabinoid agissent de façon significative via GPR119 à des concentrations physiologiques.
Ce que la pharmacologie des récepteurs orphelins raconte mal dans les titres
L’échec médiatique standard est simple : un essai positif de signalisation devient « des scientifiques ont découvert un nouveau récepteur cannabinoid ». Ce saut ignore au moins quatre filtres.
Premièrement, la dépendance à l’essai. La mobilisation du calcium, le recrutement de β-arrestine, la phosphorylation d’ERK, la redistribution dynamique de la masse et la liaison radioligand ne posent pas la même question. Un ligand lipophile peut perturber les membranes, modifier le trafic du récepteur ou montrer un biais de voie. Si le récepteur est surexprimé, les composés faibles commencent à paraître puissants.
Deuxièmement, les différences entre espèces. Le GPR55 humain n’est pas le GPR55 murin dans tous ses détails pharmacologiques, et il en va de même pour GPR18. Un profil de ligand établi dans des cellules HEK293 avec le récepteur humain ne prédira pas nécessairement une étude de douleur chez le rat.
Troisièmement, la concentration. Beaucoup d’articles sur les cannabinoid rapportent une activité micromolaire in vitro. Cela peut avoir une importance pharmacologique, mais pas automatiquement. Les niveaux tissulaires après inhalation, administration orale, métabolisme de premier passage ou accumulation locale dans la graisse et les membranes varient énormément. Une liaison in vitro n’est pas un mécanisme clinique.
Quatrièmement, le contexte. Un récepteur dans les cellules immunitaires peut médiatiser un effet ; le même récepteur dans les ostéoclastes, un autre. Ajoutez les interactions croisées avec les canaux TRP, les PPAR, les récepteurs de la sérotonine et même les canaux sodiques, et le récit propre d’un ligand/une cible s’effondre rapidement.
C’est pourquoi « CB3 » n’a jamais vraiment pris. GPR55 possède une biologie crédible dans la signalisation calcique, la douleur, le remodelage osseux et l’inflammation. Il présente aussi une pharmacologie cannabinoid contradictoire, une forte sensibilité aux essais, et une solide revendication concurrente selon laquelle les lipides de la famille LPI sont ses principaux ligands physiologiques. GPR18 est encore plus incertain. GPR119 n’appartient généralement pas à la même catégorie, si ce n’est comme rappel que les GPCR lipidiques sont faciles à associer abusivement aux cannabinoid.
Pour la science des cannabinoid, la leçon est la retenue. Ces récepteurs peuvent être très importants. Ils ne justifient simplement pas un renommage prématuré.
Signalisation de la sérotonine : là où les cannabinoid interfèrent avec les systèmes 5-HT
La sérotonine est le point où de nombreuses affirmations populaires sur CBD deviennent à la fois plus plausibles et plus glissantes. La partie plausible est simple : dans les essais cellulaires, les modèles murins d’anxiété, les paradigmes de stress et un petit nombre d’études expérimentales chez l’humain, 5-HT1A réapparaît sans cesse comme un nœud important des effets comportementaux du CBD. La partie glissante est que « agit sur la sérotonine » peut vouloir dire plusieurs choses différentes. Il peut s’agir d’un agonisme direct sur le site orthostérique. Il peut s’agir d’une modulation allostérique positive. Il peut s’agir d’une facilitation de la signalisation du récepteur sans liaison à haute affinité. Ou il peut s’agir d’une modification par CBD de l’activité du réseau en amont ou en aval des neurones sérotoninergiques, produisant un résultat dépendant de la sérotonine sans être du tout un médicament classique des récepteurs sérotoninergiques.
Cette distinction est importante. Beaucoup. Si un composé apaise un comportement d’une manière bloquée par un antagoniste 5-HT1A tel que WAY-100635, cela ne prouve pas à lui seul que le composé est un agoniste de 5-HT1A. Cela prouve une dépendance à la signalisation 5-HT1A dans ce modèle. Ce n’est pas la même chose, et les textes de vulgarisation sur le cannabis confondent souvent les deux.
5-HT1A et la question de l’anxiété
Le lien sérotoninergique le plus solide pour les cannabinoid, en particulier CBD, est 5-HT1A. Ce récepteur est un récepteur de la sérotonine couplé à Gi/o, exprimé à la fois comme auto-récepteur sur les neurones sérotoninergiques du raphé et comme récepteur postsynaptique dans des régions impliquées dans l’anxiété, notamment l’hippocampe, l’amygdale et le cortex préfrontal. Les médicaments qui activent ou recrutent ce système peuvent réduire l’anxiété dans certains contextes, mais l’emplacement du récepteur compte : diminuer le tir sérotoninergique via les auto-récepteurs n’est pas la même chose que façonner une signalisation postsynaptique dans les circuits limbiques.
CBD est entré dans cette discussion par des travaux précliniques des années 2000 et 2010 montrant des effets de type anxiolytique dans des tests comme le labyrinthe en croix surélevé, le test de conflit de Vogel et des paradigmes de peur contextuelle, avec un blocage partiel par WAY-100635. Un article souvent cité est celui de Campos et Guimarães, 2008, qui a montré qu’une administration de CBD dans le cortex prélimbique réduisait les réponses cardiovasculaires liées au stress de contention et que des mécanismes 5-HT1A contribuaient à l’effet. Une autre étude humaine importante est celle de Bergamaschi et collègues, 2011 : dans un test de prise de parole en public simulée, 600 mg de CBD oral ont réduit l’anxiété chez des sujets souffrant de trouble d’anxiété sociale par rapport au placebo. Cet article ne prouvait pas la médiation par 5-HT1A chez l’humain, mais il concordait avec le schéma préclinique et a aidé à faire de la sérotonine un mécanisme candidat sérieux plutôt qu’une formule marketing.
La pharmacologie du récepteur n’a toutefois jamais abouti à une simple histoire du type « CBD est un agoniste sérotoninergique ». Des travaux in vitro précoces suggéraient que CBD pouvait déplacer des ligands au niveau des récepteurs humains 5-HT1A et agir comme agoniste dans certains essais de signalisation, mais les affinités étaient modestes et dépendantes de l’essai. Russo et collègues ont rapporté en 2005 que CBD agissait comme agoniste des récepteurs humains clonés 5-HT1A dans des essais de liaison [35S]GTPγS. Cette observation a été influente, mais des travaux ultérieurs l’ont compliquée. Certains groupes ont vu une activité directe faible. D’autres ont observé une facilitation fonctionnelle mieux expliquée par des effets allostériques ou membranaires. La littérature n’est cohérente que sur un point : 5-HT1A compte davantage pour la pharmacologie anxiolytique du CBD que CB1 ou CB2 seuls ne peuvent l’expliquer.
C’est pourquoi le réductionnisme récepteur échoue. Si CBD n’était qu’un agoniste 5-HT1A pur et propre, son profil ressemblerait plus nettement à celui des anxiolytiques sérotoninergiques connus. Or le signal comportemental est très dépendant du contexte, montrant souvent des courbes dose-réponse en U inversé. Dans certains tests chez le rongeur, des doses modérées réduisent les comportements de type anxieux alors que des doses plus faibles ou plus élevées font moins. C’est un signal d’alarme contre les récits mono-récepteurs. L’activation de TRPV1 à des concentrations plus élevées est l’une des explications proposées. Il en va de même pour les effets sur le tonus endocannabinoïde, la recapture de l’adénosine et la gestion du calcium intracellulaire. Une molécule peut recruter 5-HT1A tout en refusant d’agir comme un médicament sérotoninergique classique.
Liaison directe versus effets sérotoninergiques indirects
La meilleure manière de lire les preuves sur la sérotonine est par paliers. Au niveau moléculaire, il existe un soutien pour une interaction directe entre CBD et 5-HT1A, mais pas de ce type d’interaction nette, à forte affinité et à forte efficacité qui règle la question. Selon le système d’essai, CBD a été décrit comme agoniste faible, agoniste partiel ou modulateur allostérique positif. Ce désaccord n’est pas une simple querelle sémantique. Les agonistes orthostériques occupent le site principal de liaison à la sérotonine. Les modulateurs allostériques positifs modifient le comportement du récepteur à partir d’un autre site et peuvent amplifier les réponses endogènes à la sérotonine sans activer fortement le récepteur par eux-mêmes. Ces mécanismes n’ont pas les mêmes implications pour la dose, le timing, les effets indésirables et la traduction chez l’humain.
Les données de signalisation cellulaire pointent souvent vers une facilitation plutôt qu’une activation brutale. Dans certaines préparations, CBD renforce les cascades de signalisation médiées par 5-HT1A, y compris des effets sur ERK et d’autres voies en aval, davantage que ce que sa faible liaison laisserait prévoir. Plusieurs explications sont possibles. CBD est très lipophile et se partitionne dans les membranes, où il peut modifier l’environnement du récepteur et le couplage à la protéine G. Il peut aussi augmenter indirectement la signalisation d’anandamide, et les interactions croisée endocannabinoïde-sérotonine dans le raphé dorsal et le cerveau antérieur sont bien documentées. Il y a ensuite l’adénosine : CBD inhibe l’activité des transporteurs de nucléosides équilibratifs dans certains systèmes, augmentant l’adénosine extracellulaire et modifiant l’excitabilité neuronale de manière pouvant alimenter les circuits sérotoninergiques. Rien de tout cela ne rend 5-HT1A inutile. Cela le rend intégré.
La pharmacologie animale fournit des preuves plus solides de la dépendance à la sérotonine que de l’agonisme direct. À maintes reprises, WAY-100635 atténue les effets du CBD dans les modèles d’anxiété, de panique, de nausée et de stress. Resstel et collègues, en 2009, par exemple, ont relié l’atténuation par CBD des réponses aiguës au stress de contention à des mécanismes 5-HT1A. Les travaux de Rock et Parker sur la nausée et la nausée anticipatoire chez le rongeur ont également impliqué 5-HT1A dans le profil antiémétique de CBD. Ce sont des résultats utiles, mais ils doivent être lus comme des preuves de voie. Si le blocage de 5-HT1A supprime l’effet, la voie est impliquée. Cela ne règle pas la question de savoir si le récepteur est directement lié, modulé allostériquement, ou recruté via des modifications au niveau des circuits.
Les preuves humaines restent modestes. L’étude de Bergamaschi de 2011 est souvent citée parce qu’elle a montré un signal anxiolytique mesurable dans l’anxiété sociale lors de la prise de parole en public. Des études d’imagerie plus petites ont rapporté que CBD modifie l’activation limbique et paralimbique pendant des tâches de traitement émotionnel. Pourtant, aucune de ces études n’a identifié l’occupation du récepteur 5-HT1A chez l’humain comme peuvent le faire les études PET pour les médicaments sérotoninergiques établis. Cette absence est importante. Nous inférons le mécanisme par convergence, sans le mesurer directement aux doses cliniques.
Les effets apaisants du CBD dépendent en partie de la signalisation 5-HT1A.Limited evidence
Pourquoi le profil calmant de CBD résiste aux étiquettes réceptrices simples
CBD possède déjà un usage approuvé par la FDA, et il ne s’agit pas de l’anxiété. La notice 2024 de la solution orale de cannabidiol limite l’indication aux crises associées au syndrome de Lennox-Gastaut, au syndrome de Dravet ou au complexe de sclérose tubéreuse chez les patients âgés d’un an et plus. Ce fait constitue un bon garde-fou contre les exagérations. Un composé peut avoir des signaux anxiolytiques crédibles sans que son efficacité anxieuse soit établie au niveau réglementaire, et il peut impliquer la sérotonine sans s’inscrire proprement dans la catégorie des médicaments sérotoninergiques.
Une partie du problème tient à l’échelle. In vitro, les cannabinoid sont pharmacologiquement complexes. In vivo, ils sont encore plus complexes parce que la distribution, le métabolisme, l’accumulation tissulaire et les différences d’espèce modifient les cibles qui comptent. Un effet récepteur observé à 10 micromolaires dans des cellules transfectées peut être sans pertinence après une administration orale ordinaire, tandis qu’un effet apparemment plus faible in vitro peut compter si le composé se concentre dans un tissu cérébral riche en lipides ou si des métabolites actifs contribuent à l’effet. C’est l’une des raisons pour lesquelles les titres sur « le récepteur sérotoninergique que CBD touche » dépassent souvent les données.
Une autre raison est la biologie des circuits. L’anxiété n’est pas générée par un seul récepteur. Elle émerge d’interactions entre l’amygdale, le noyau du lit de la strie terminale, le cortex préfrontal médian, l’hippocampe, l’hypothalamus et des noyaux du tronc cérébral, dont le raphé dorsal. CBD semble modifier l’activité à travers ce réseau. Une partie de ce changement recrute probablement 5-HT1A. Une autre peut impliquer TRPV1, qui peut s’opposer à l’anxiolyse à des doses plus élevées. Une autre encore peut reposer sur des modifications du tonus d’anandamide liées à FAAH, bien que l’inhibition de FAAH par CBD chez l’humain aux expositions thérapeutiques soit débattue. Certaines peuvent refléter des effets anti-inflammatoires ou autonomes qui rétroagissent sur l’anxiété ressentie. Une fois cette vision en réseau adoptée, l’échec d’une explication à étiquette unique cesse d’être une faiblesse et devient un compte rendu réaliste de la pharmacologie.
C’est aussi là que se dirige le développement de médicaments. L’ère de la chimie médicinale s’intéresse moins à savoir si un composé est « comme THC » qu’à définir des combinaisons de cibles et à séparer les effets souhaités de l’intoxication. Cette logique apparaît dans des travaux très éloignés de la sérotonine, depuis le criblage structurel de CB2 dans l’article de 2016 du Journal of Medicinal Chemistry « Library Docking for Cannabinoid-2 Receptor Ligands » jusqu’aux efforts plus récents pour dissocier l’analgésie de l’altération centrale. Elle apparaît aussi dans les programmes anxiolytiques à l’étape des entreprises. En 2025, MIRA Pharmaceuticals a déclaré que son candidat MIRA-55 présentait un « differentiated mechanism of action » et une « anxiolytic activity relative to THC » dans des données précliniques rapportées dans un communiqué Nasdaq. Le niveau de preuve doit rester explicite ici : préclinique, rapporté par l’entreprise, pas une preuve clinique. Néanmoins, le signal est significatif comme indicateur de marché et de recherche. Les entreprises cherchent activement des agents inspirés des cannabinoid capables d’apaiser sans agir comme THC, et les mécanismes tournés vers la sérotonine font partie de cette recherche.
Le contexte de santé publique en fait plus qu’un débat académique. En 2025, HHS a déclaré que 7-hydroxymitragynine « poses an imminent hazard to public safety » en soutenant une action de classement contre des produits 7-OH renforcés dangereux. Différentes modifications chimiques créent différents profils de cibles et différents risques. La même leçon s’applique à l’espace des cannabinoid. Si un produit est traité comme interchangeable avec des cannabinoid végétaux familiers parce qu’il sonne adjacent à THC ou CBD, la pharmacologie est aplatie et l’évaluation de la sécurité en pâtit.
Alors, où se situe la preuve ? 5-HT1A est le mécanisme sérotoninergique le mieux étayé pour les effets calmants de CBD, mais l’affirmation la plus solide que les données soutiennent actuellement n’est pas « CBD est un agoniste de la sérotonine ». Elle est plus étroite et plus défendable : CBD produit souvent des effets de type anxiolytique et de tampon du stress qui dépendent en partie de la signalisation 5-HT1A, tandis que le mode exact d’engagement semble varier selon l’essai, la dose, le tissu et le contexte des circuits. C’est peut-être moins net qu’un slogan à récepteur unique. C’est aussi beaucoup plus proche de la vérité.
Au-delà de la liste demandée : les canaux sodiques et d’autres cibles non canoniques qui modifient déjà la conversation sur la douleur
Pendant des années, la plupart des discussions publiques sur la pharmacologie cannabinoid de la douleur sont restées prisonnières d’un récit à deux récepteurs : CB1 explique les effets psychoactifs, CB2 les effets immunitaires, et tout le reste est secondaire. Ce cadre est désormais trop étroit. Même dans le champ plus restreint de la douleur, les cannabinoid ne se contentent pas de toucher les canaux TRP, les PPAR, les GPCR orphelins ou les voies liées à la sérotonine. Ils interagissent aussi avec les canaux sodiques voltage-dépendants, au cœur de l’excitabilité des nocicepteurs. C’est important parce que NaV1.7 et NaV1.8 ne sont pas des notes de bas de page périphériques ; ils font partie des portes moléculaires les plus étudiées de la signalisation douloureuse dans les neurones sensoriels de petit diamètre.
Le changement dépasse le cadre académique. Les développeurs de médicaments ont passé des années à tenter de bloquer la transmission de la douleur au niveau des nerfs périphériques sans reproduire la sédation, l’intoxication, les troubles de la mémoire et le potentiel d’abus associés à une forte activation centrale de CB1. Si un cannabinoid, ou un squelette dérivé des cannabinoid, peut atténuer le tir des nocicepteurs en agissant sur des canaux NaV hors du cerveau, cela ouvre une logique thérapeutique très différente. On passe de « À quel point cela touche-t-il CB1 ? » à « Où agit-il, à quelle concentration, et dans quel tissu ? »
Cette carte des cibles plus large s’inscrit aussi dans le moment réglementaire actuel. En 2025, le U.S. Department of Health and Human Services a averti que « 7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety », rappelant que de petits changements chimiques peuvent produire des pharmacologies et des profils de sécurité très différents. Les politiques relatives au cannabis ont souvent pris du retard par rapport à ce fait élémentaire. Traiter tous les composés adjacents aux intoxicants comme s’ils ne différaient que par leur source ou leur puissance équivalente au THC manque l’essentiel. C’est la pharmacologie au niveau des cibles qui prédit l’effet, le risque et le potentiel médicamenteux.
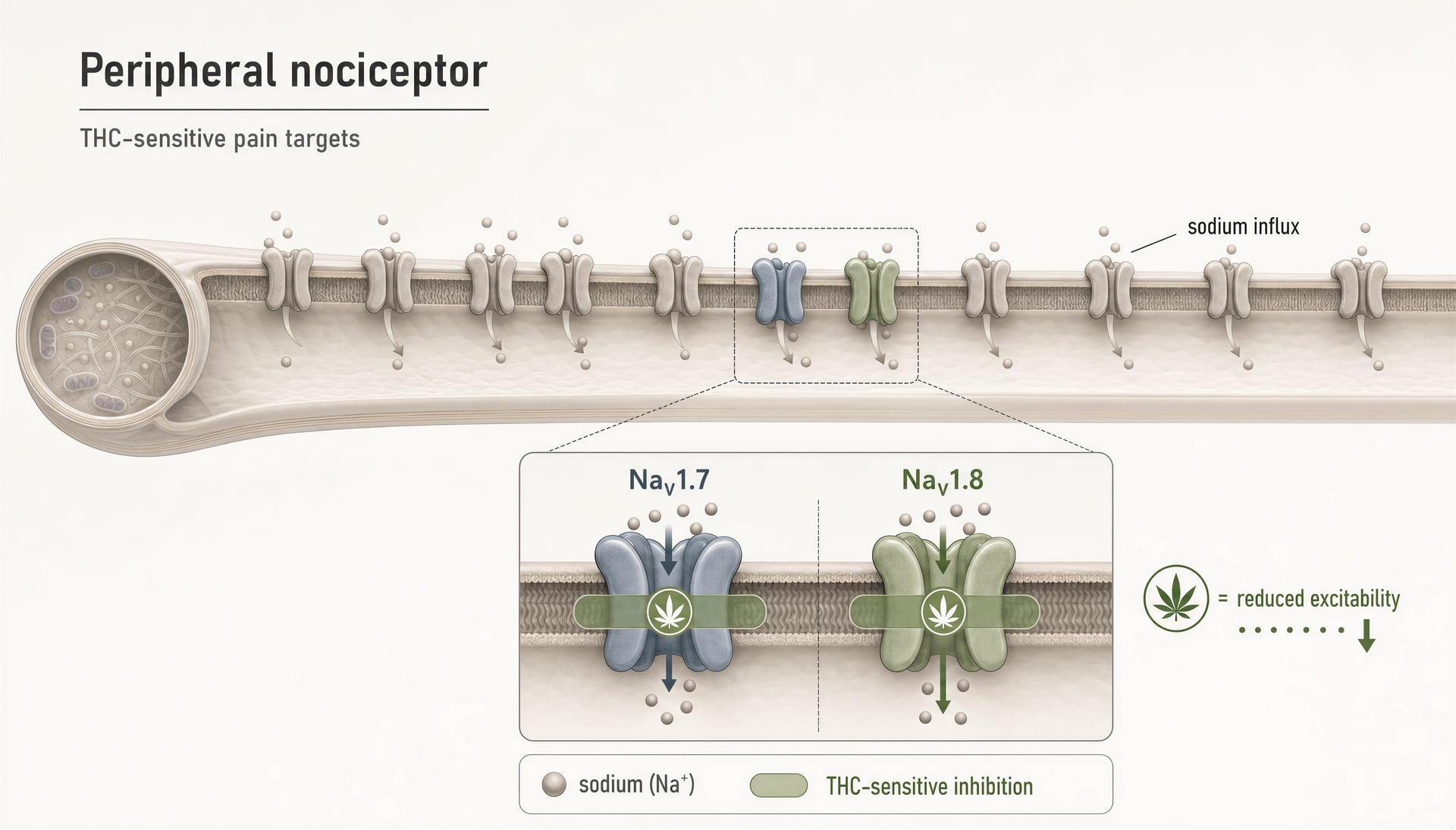
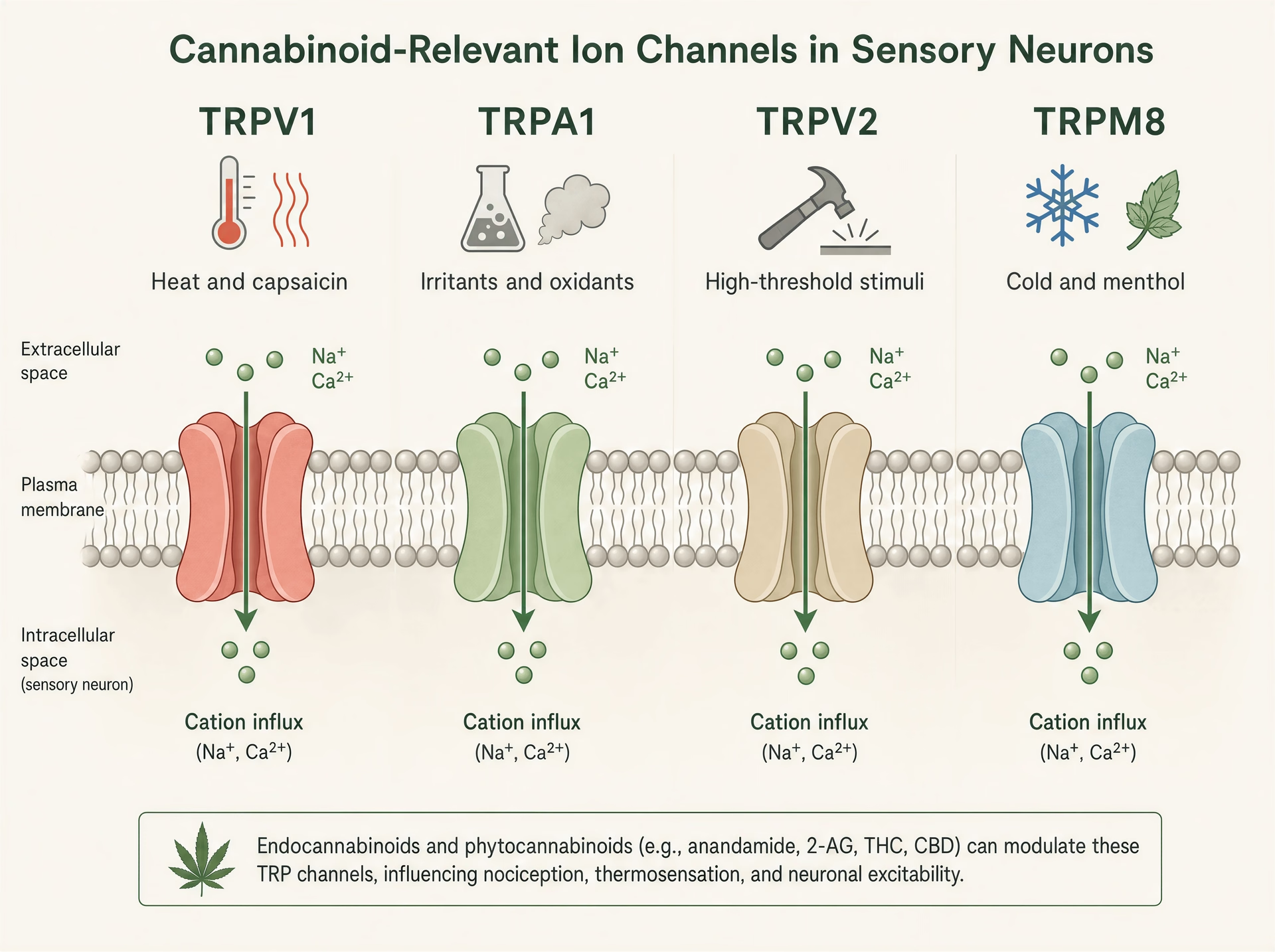{kind=link}
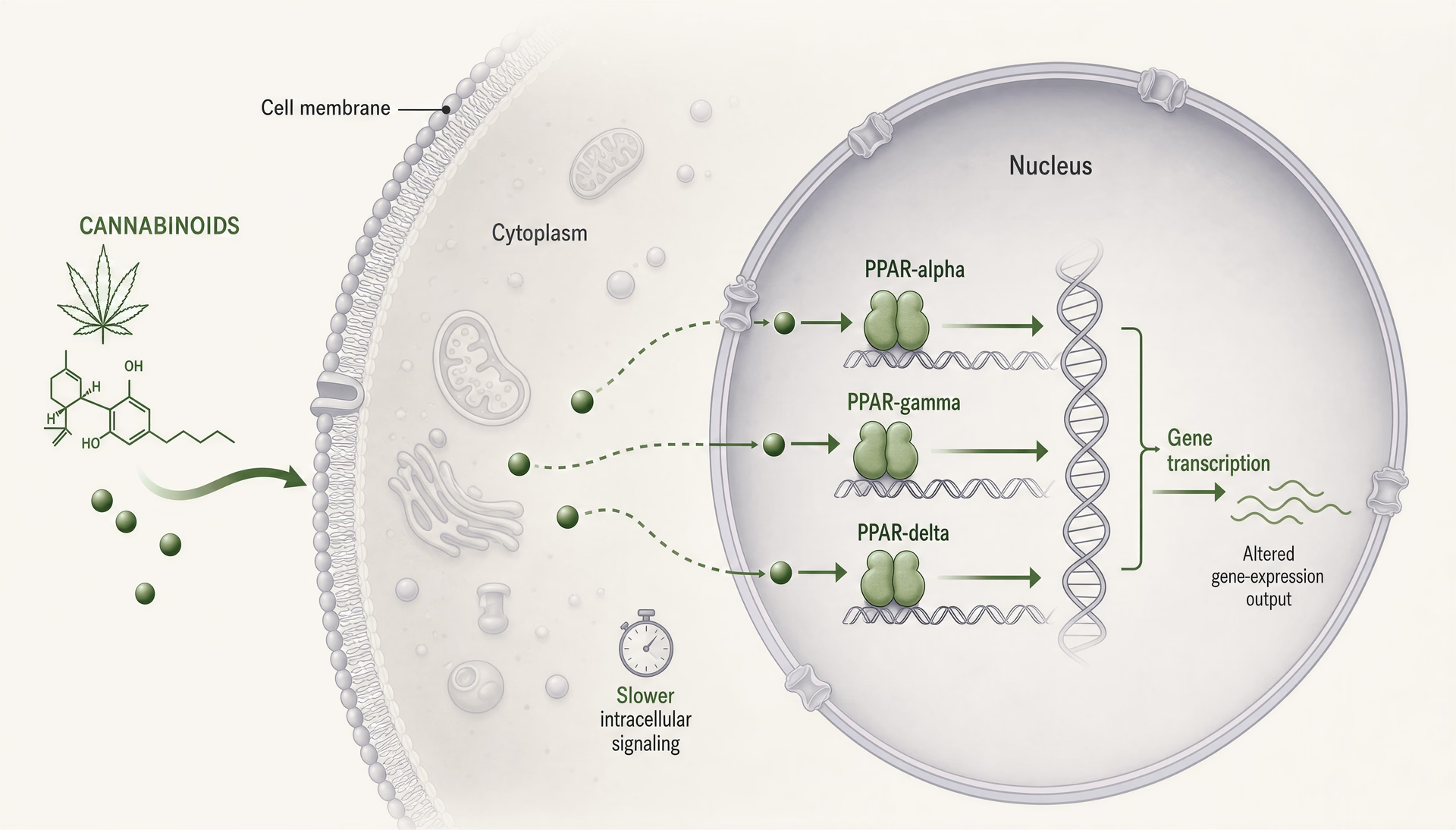{kind=link}
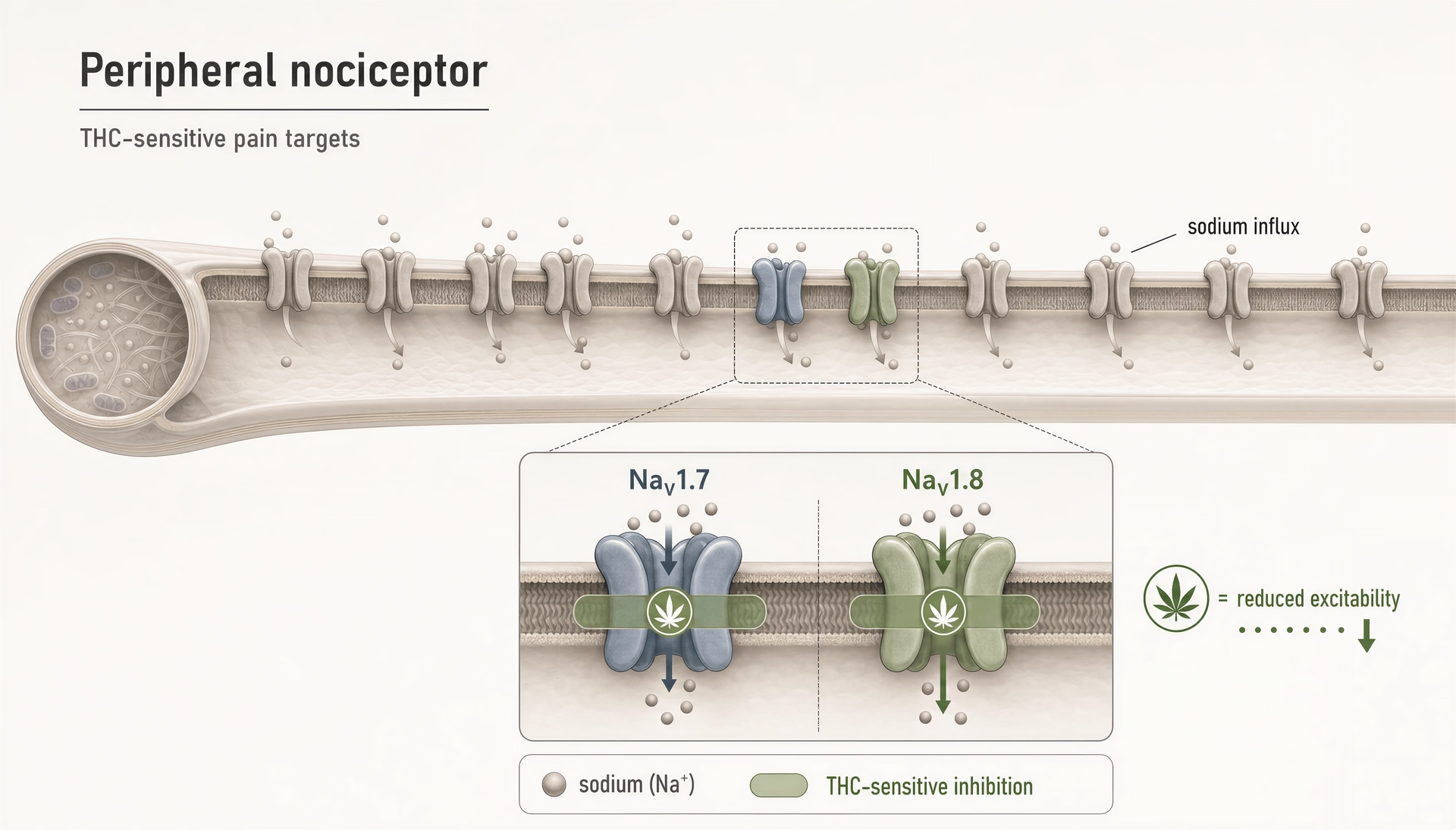{kind=link}
Le THC inhibe les nocicepteurs périphériques en ciblant les canaux sodiques NaV1.7 et NaV1.8.Preliminary evidence
THC sur les canaux nociceptifs périphériques NaV1.7 et NaV1.8
La raison la plus directe pour laquelle les canaux sodiques doivent désormais figurer dans toute carte sérieuse des cannabinoid est le rapport 2025 du groupe de l’Université hébraïque de Jérusalem montrant que THC inhibe les nocicepteurs périphériques en ciblant les « NaV1.7 and NaV1.8 nociceptive sodium channels ». C’est une extension significative du vocabulaire du domaine. NaV1.7 et NaV1.8 sont fortement exprimés dans les neurones périphériques sensibles à la douleur, et leur rôle dans la biologie humaine de la douleur n’a rien de spéculatif. Les mutations perte de fonction de NaV1.7 peuvent provoquer une insensibilité congénitale à la douleur ; les mutations gain de fonction peuvent déclencher des syndromes douloureux sévères. NaV1.8 est lui aussi lié aux états douloureux inflammatoires et neuropathiques, car il soutient le tir répétitif des nocicepteurs, en particulier dans des conditions de dépolarisation.
Ainsi, lorsqu’il est démontré que THC inhibe ces canaux, la découverte n’entre pas dans la case des « effets hors cible divers ». Elle pointe vers un mécanisme capable de réduire directement l’excitabilité des fibres nociceptives avant que les signaux n’atteignent la moelle épinière ou le cerveau.
C’est une classe mécanistique différente des récits cannabinoid les plus connus. TRPV1, reconnu dans les travaux ayant contribué à la part de David Julius dans le prix Nobel 2021, peut être activé ou désensibilisé par plusieurs cannabinoid, dont CBD et CBG, avec des effets très dépendants de la dose et du timing. La signalisation PPAR-gamma a été invoquée pour des effets anti-inflammatoires et métaboliques, souvent compliqués par le fait que l’accumulation intracellulaire et les métabolites peuvent compter autant que les composés parents. GPR55 reste suffisamment débattu pour que l’appeler « CB3 » relève encore davantage du slogan que de la science établie. Les liens avec la sérotonine, surtout 5-HT1A, aident à expliquer une partie du profil anxiolytique de CBD, mais les circuits sont dépendants du contexte et souvent indirects. L’inhibition des canaux sodiques est moins glamour. Elle est aussi, pour la douleur, potentiellement plus pratique.
Un point clé est la promiscuité pharmacologique. Les cannabinoid sont souvent des ligands « sales » au sens technique : ils engagent plusieurs cibles avec des affinités et des conséquences fonctionnelles différentes. Ce n’est pas un défaut de la science ; c’est la science elle-même. THC reste peut-être surtout connu pour son agonisme central de CB1, mais cela n’annule pas sa capacité à moduler des canaux ioniques périphériques dans les bonnes conditions. La vraie question est de savoir si ces conditions sont atteignables in vivo d’une manière qui aide les patients plus qu’elle ne les nuit. La découverte de l’Université hébraïque montre que c’est au moins suffisamment plausible pour mériter une attention sérieuse en développement de médicaments.
Un analgésique cannabinoïde peut soulager la douleur sans produire l’euphorie via des mécanismes périphériques ou non-CB1.Preliminary evidence
Analgésie périphérique sans intoxication centrale
C’est là que le champ de la douleur devient intéressant. Un mécanisme cannabinoid qui réduit le tir des nocicepteurs périphériques pourrait, en théorie du moins, séparer l’analgésie de la baisse cognitive généralement liée à l’activation de CB1 dans le cerveau. Cette distinction est au cœur du travail translationnel actuel, pas un bénéfice secondaire.
Le rapport ScienceDaily de 2026 a exprimé l’idée en langage simple : des chercheurs ont identifié « a cannabis compound that relieves pain without the high ». Cette formulation doit être lue avec prudence. Il s’agit d’un signal au stade de la recherche, pas d’une thérapie établie, et les résumés populaires simplifient souvent les détails mécanistiques. Néanmoins, l’importance translationnelle est évidente. Si une activité analgésique peut être obtenue via une restriction périphérique, une pénétration cérébrale limitée, un engagement sélectif de cibles non CB1, ou une combinaison des trois, alors l’ancien compromis entre soulagement de la douleur et intoxication n’est pas figé par la nature. C’est un problème de chimie médicinale.
Ce point aide aussi à expliquer pourquoi le domaine a dépassé les étiquettes grossières des récepteurs. L’article de 2016 de l’ACS Journal of Medicinal Chemistry, « Library Docking for Cannabinoid-2 Receptor Ligands », reflète un virage plus large vers la conception fondée sur la structure plutôt que le traitement des cannabinoid comme une famille pharmacologique unique avec un seul axe utile de variation. Les chimistes cherchent désormais comment ajuster la forme du squelette, la lipophilie, le biais récepteur, la distribution tissulaire et le devenir métabolique. L’objectif n’est pas seulement une activité plus forte. L’objectif est une activité sélective au bon endroit.
L’analgésie périphérique est précisément le type d’issue où ces distinctions comptent. Un composé qui traverse mal la barrière hémato-encéphalique, mais inhibe de manière significative NaV1.7 ou NaV1.8 dans les nocicepteurs, pourrait soulager la douleur inflammatoire ou neuropathique avec beaucoup moins d’intoxication que THC lui-même. Cela reste une ambition, pas un fait clinique. Pourtant, les travaux de l’Université hébraïque donnent à cette ambition un ancrage moléculaire.[8]Agriculture Improvement Act of 2018. U.S. Congress. Congress.gov, 2018. https://www.congress.gov/bill/115th-congress/house-bill/2/text
Ils précisent aussi notre lecture des produits à base de cannabis déjà en circulation. La définition légale du hemp dans le Farm Bill de 2018 repose sur une teneur en delta-9 THC « not more than 0.3 percent on a dry weight basis ». Ce chiffre est réglementaire, pas pharmacologique. Il ne dit rien des canaux sodiques, de l’activation des TRP, de la signalisation 5-HT1A, des métabolites actifs ou de l’exposition tissulaire. Il en va de même pour les nouveaux intoxicants renforcés ou semi-synthétiques. La sécurité ne peut pas être déduite des histoires d’origine. Elle doit être déduite des cibles, des concentrations et de la vraie pharmacocinétique.
Pourquoi ces résultats comptent pour les futurs médicaments à base de cannabinoid
L’implication la plus forte du récit NaV1.7/NaV1.8 est que les futurs médicaments cannabinoid pourraient réussir précisément en étant moins « cannabinoid-like » au sens populaire. Autrement dit, les descendants utiles de la chimie du cannabis ne seront peut-être pas des médicaments qui miment largement le THC fumé. Ils peuvent être des composés qui empruntent une partie du squelette, évitent la signalisation centrale de CB1 et agissent à la place sur des canaux ioniques périphériques ou des ensembles de cibles non canoniques mixtes.
Cette possibilité s’inscrit déjà dans le socle de preuves plus large. L’usage approuvé du CBD concerne les syndromes épileptiques, pas l’analgésie de routine, et même là, sa pharmacologie ne s’explique pas bien par CB1 ou CB2 seuls. La notice de la solution orale de cannabidiol indique qu’elle est destinée aux crises associées au syndrome de Lennox-Gastaut, au syndrome de Dravet ou au complexe de sclérose tubéreuse chez les patients âgés d’un an et plus. En d’autres termes, le seul grand médicament cannabinoid approuvé par la FDA en usage large aujourd’hui résiste déjà au récit récepteur simpliste. Le champ de la douleur rattrape la même leçon.
Les premiers programmes des entreprises vont dans le même sens, même s’ils doivent être traités avec prudence. En 2025, MIRA Pharmaceuticals a rapporté des données précliniques affirmant que MIRA-55 présentait un « differentiated mechanism of action » et une « anxiolytic activity relative to THC ». Les communiqués d’entreprise ne constituent pas une preuve neutre, et les signaux précliniques échouent souvent. Même ainsi, ils montrent où va la chimie médicinale : s’éloigner de l’imitation non dirigée du THC et aller vers une conception façonnée par le mécanisme.
Pour la douleur, les canaux sodiques pourraient devenir l’une des branches les plus importantes de cette stratégie. Pas la seule. TRPV1, TRPA1, PPAR-gamma, GPR55, les voies liées à l’adénosine et la modulation sérotoninergique resteront dans le tableau. Mais NaV1.7 et NaV1.8 apportent quelque chose de particulièrement attrayant : une connexion directe avec le comportement électrique des fibres douloureuses périphériques. Cela en fait des cibles d’une concrétude inhabituelle dans un domaine saturé d’explications indirectes.
Le résultat est une manière plus claire de penser les cannabinoid et la douleur. Non pas CB1 contre CB2. Non pas végétal contre synthétique. Et non pas « high » contre « medical » comme s’il s’agissait de catégories moléculaires. La meilleure distinction est celle entre mécanismes d’intoxication centrale et mécanismes potentiellement utiles en périphérie. La découverte de l’Université hébraïque place THC lui-même des deux côtés de cette ligne. C’est précisément pour cela qu’elle importe.
| Cannabinoïde | Cibles non-CB1/CB2 mises en avant dans l’article | Principale mise en garde |
|---|---|---|
| CBD | TRPV1, TRPA1, TRPM8, 5-HT1A, PPAR-gamma, GPR55, signalisation liée à l’adénosine | De nombreux signaux proviennent d’études dépendantes des essais et souvent micromolaires |
| THC | TRPV2, interactions TRPV1/TRPA1 rapportées, NaV1.7, NaV1.8 | Les effets centraux de CB1 peuvent dominer et masquer d’autres mécanismes |
| CBG | TRPA1, TRPV1, TRPM8, interactions liées à alpha-2 adrénergique et à 5-HT1A | La transposition de la concentration in vitro à l’exposition humaine est incertaine |
| CBC | TRPA1, canaux de la famille TRPV | Les preuves chez l’humain sont limitées |
| THCV | Possibilités non-CB1 incluant TRP et la signalisation métabolique | La dose et le contexte peuvent modifier le comportement apparent |
| CBDA / THCA | Actions liées à 5-HT1A, aux TRP et aux enzymes discutées dans des travaux précliniques | La stabilité, la décarboxylation et l’exposition compliquent l’interprétation |
En quoi les cannabinoid spécifiques diffèrent lorsque l’on ne s’interroge plus seulement sur CB1 et CB2
Dès lors qu’on cesse de traiter CB1 et CB2 comme l’ensemble de l’histoire, la liste familière des cannabinoid paraît beaucoup moins ordonnée. Ces molécules ne sont pas des clés propres pour deux serrures. Ce sont des composés lipophiles, sensibles à la concentration, capables de toucher des canaux ioniques, des récepteurs nucléaires, des processus de transport, des GPCR orphelins et, dans certains cas, des canaux sodiques voltage-dépendants. Cela compte parce que la douleur, l’inflammation, le contrôle des crises, l’anxiété et les effets indésirables se superposent souvent mal à de simples étiquettes « agoniste de CB1 » ou « agoniste de CB2 ».
CBD a imposé ce changement plus que tout autre cannabinoid. Sa faible efficacité classique sur les récepteurs cannabinoid a rendu l’ancien cadre difficile à défendre, surtout après qu’une solution orale de cannabidiol approuvée par la FDA a obtenu des indications pour les crises associées au syndrome de Lennox-Gastaut, au syndrome de Dravet et au complexe de sclérose tubéreuse chez les patients âgés d’un an et plus. Un cannabinoid cliniquement utile avec une intoxication de type CB1 limitée posait problème au réductionnisme récepteur. Les chercheurs ont dû regarder ailleurs.
Le champ plus large a suivi. Les travaux sur les canaux TRP se sont appuyés sur une biologie sensorielle reconnue au plus haut niveau lorsque le prix Nobel de physiologie ou médecine 2021 a été décerné à David Julius et Ardem Patapoutian pour leurs découvertes sur les récepteurs de la température et du toucher. La pharmacologie des cannabinoid croise directement cette biologie : TRPV1, TRPA1 et TRPM8 reviennent sans cesse parce que de nombreux phytocannabinoid peuvent les activer, les inhiber ou les désensibiliser à des concentrations expérimentalement pertinentes. Cela ne signifie pas que chaque résultat d’essai prédit un effet humain. Cela signifie que l’ancienne idée selon laquelle l’action « réelle » des cannabinoid commence et s’arrête à CB1/CB2 est fausse.
CBD : le prototype de la complexité hors CB1/CB2
CBD est le meilleur exemple de l’importance de la promiscuité des cibles. Il a une faible affinité et une efficacité limitée sur CB1 et CB2 par rapport au THC, mais il fait clairement quelque chose d’important sur le plan biologique. L’écart entre ces deux faits a produit une série d’hypothèses de travail, certaines plus solides que d’autres.
Les canaux TRP ont compté parmi les premières alternatives sérieuses. CBD active TRPV1 dans des systèmes hétérologues, et TRPV1 n’est pas une voie secondaire obscure ; c’est un canal central de la nociception et de la douleur inflammatoire. L’activation peut sembler contre-intuitive si l’objectif thérapeutique est l’analgésie, mais une activation répétée ou soutenue de TRPV1 produit souvent une désensibilisation, réduisant l’excitabilité ultérieure. C’est l’une des raisons pour lesquelles la pharmacologie des TRP peut paraître contradictoire sur le papier. Un composé peut d’abord activer puis calmer le système. CBD montre aussi des actions sur TRPA1 et peut inhiber TRPM8 dans certains modèles, ce qui le rend plus large sur le plan pharmacologique que le résumé public habituel du « cannabinoid non intoxicant ».
L’histoire de la sérotonine est plus contestée mais toujours importante. Une large littérature préclinique relie CBD à des effets dépendant de 5-HT1A, surtout dans les modèles d’anxiété, de stress et de nausée. L’affirmation la plus nette n’est pas que CBD soit un agoniste 5-HT1A simple et à forte affinité à la manière dont on décrit les pharmacologies liées au buspirone, mais que la signalisation 5-HT1A contribue souvent aux effets de CBD in vivo, parfois via un agonisme partiel, parfois par des mécanismes allostériques ou de circuit encore non résolus. Cette distinction importe. Trop de résumés réduisent « implique 5-HT1A » à « agit par la sérotonine ». Les données ne soutiennent pas cette simplification.
PPAR-gamma est un autre candidat récurrent, et ici la chimie intracellulaire compte. Les PPAR sont des récepteurs nucléaires ; une molécule lipophile qui se partitionne dans les membranes et les cellules peut donc les affecter d’une manière qu’un modèle de récepteur de surface ne voit pas. CBD a été rapporté comme capable d’activer PPAR-gamma dans des systèmes cellulaires, et la signalisation PPAR-gamma a des liens plausibles avec l’inflammation, le métabolisme lipidique, la fibrose et la neuroinflammation. Mais il y a un piège : certains effets liés aux PPAR peuvent refléter des métabolites, une exposition plus longue ou des changements indirects dans les médiateurs lipidiques endogènes plutôt qu’une occupation réceptrice en une seule étape. La pharmacologie est suffisamment réelle pour être étudiée sérieusement, mais moins bonne comme slogan que comme mécanisme.
La signalisation adénosinergique est entrée dans le tableau parce que CBD semble inhiber le transport des nucléosides équilibratifs dans certains systèmes, augmentant potentiellement le tonus extracellulaire d’adénosine et influençant indirectement les voies anti-inflammatoires liées à A2A. Là encore, il ne s’agit pas d’une pharmacologie de liaison récepteur propre. C’est de la pharmacologie des transporteurs et du contexte tissulaire. Si cela paraît plus désordonné que « CBD touche CB2 », c’est parce que cela l’est. C’est aussi plus plausible.
Puis vient GPR55, souvent présenté comme un « CB3 » putatif. Cette étiquette est encore trop confiante. CBD peut antagoniser ou moduler autrement la signalisation liée à GPR55 dans plusieurs systèmes expérimentaux, et GPR55 a été impliqué dans l’excitabilité, la biologie osseuse, l’inflammation et des voies liées au cancer. Mais les ligands endogènes du récepteur, son couplage aux voies et sa pertinence translationnelle restent débattus. GPR55 est un générateur d’hypothèses utile, pas un remplaçant établi de CB1/CB2.
En résumé, CBD est devenu le prototype de la pharmacologie cannabinoid hors CB1/CB2 parce que ses effets cliniquement pertinents ne pouvaient pas être expliqués autrement. C’est toujours vrai.
CBG, CBC, THCV, cannabinoid acides et pharmacologie des minor cannabinoid
Les minor cannabinoid sont souvent vendus au public comme si chacun avait un trait de personnalité unique. La pharmacologie n’oblige pas à coopérer.
CBG est généralement décrit comme faiblement actif sur CB1 et CB2, mais les signaux les plus intéressants se situent en dehors de ces récepteurs. Il interagit avec les systèmes adrénergiques alpha-2 et 5-HT1A dans certains essais, montre une activité sur les canaux TRP et a été étudié pour des effets anti-inflammatoires et analgésiques qui ne se réduisent pas proprement à l’activation classique des récepteurs cannabinoid. Il illustre aussi un problème récurrent : une activité in vitro micromolaire est facile à publier et difficile à traduire. Une interaction à 10 micromolaires peut compter dans une boîte et pas dans le plasma humain, ou ne compter que dans des tissus où le composé se concentre.
CBC est longtemps resté sous-caractérisé par rapport à l’engouement qui l’entoure. Il semble engager plus convaincument les canaux de la famille TRPA1 et TRPV que CB1, et certaines études suggèrent des effets anti-inflammatoires ou de type analgésique chez l’animal. Il existe aussi un intérêt pour son interaction avec le tonus endocannabinoïde, y compris des effets susceptibles de modifier indirectement la signalisation de l’anandamide. Mais « CBC agit via les canaux TRP » reste un point de départ, pas une réponse finale. Les preuves humaines sont très maigres.
THCV est un cas plus complexe, car il peut se comporter différemment selon la dose et le contexte même au niveau de CB1, étant souvent décrit comme antagoniste neutre ou ligand de faible efficacité à certaines concentrations et comme composé plus agoniste à d’autres. En dehors de CB1/CB2, THCV a été relié à des actions TRP et à des effets métaboliques, avec un intérêt répété pour l’appétit, le contrôle glycémique et l’équilibre énergétique. Une partie de cet enthousiasme a dépassé les preuves depuis des années. La meilleure interprétation est que THCV est pharmacologiquement intéressant précisément parce qu’il ne suit pas le modèle du THC, et non parce qu’une seule cible secondaire a complètement expliqué son profil.
Les cannabinoid acides méritent davantage d’attention qu’ils n’en reçoivent. THCA et CBDA sont souvent traités comme de simples précurseurs « bruts », mais leur pharmacologie n’est pas simplement inactive en attente de décarboxylation. CBDA possède certaines des données les plus intrigantes concernant des effets anti-nausée liés à 5-HT1A dans les travaux précliniques, et les deux cannabinoid acides ont montré in vitro des actions sur les TRP et les enzymes. Leur pénétration cérébrale plus faible que celle des cannabinoid neutres peut même être un avantage pour certaines cibles périphériques ou gastro-intestinales. Le problème est la qualité des données et le réalisme des doses. De nombreuses affirmations reposent sur des études animales rares ou des essais cellulaires à pertinence humaine incertaine.
C’est ici que les politiques actuelles et la chimie des produits entrent en collision avec la pharmacologie. Le Farm Bill de 2018 a défini le hemp à l’aide d’un seuil de delta-9 THC « not more than 0.3 percent on a dry weight basis ». Cette limite juridique ne dit rien de GPR55, de TRPV1, de NaV1.7, des métabolites ou des analogues semi-synthétiques. Les régulateurs ont commencé à réagir à ce décalage dans des domaines voisins. En 2025, HHS a déclaré que « 7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety » en soutenant une action de la DEA sur des produits 7-OH renforcés. Classe de médicament différente, même leçon : les produits modifiés ou enrichis peuvent avoir des profils de sécurité très éloignés du matériel source. Les cannabinoid n’échappent pas à cette logique.
Métabolites, allégations d’entourage et problème chimique
L’une des raisons pour lesquelles la pharmacologie cannabinoid devient glissante est que le composé parent ne représente pas toujours toute l’exposition. Les métabolites peuvent compter, parfois beaucoup. Le public l’a surtout appris à propos des métabolites intoxicants du THC, mais le principe général s’applique à toute la science des cannabinoid. La voie d’administration, le métabolisme de premier passage, le partitionnement tissulaire et les différences d’espèce peuvent tous modifier les cibles réellement engagées.
Même THC, le composé le plus étroitement associé à CB1, n’est pas limité à la biologie CB1. En 2025, des chercheurs de l’Université hébraïque ont rapporté que THC inhibe les nocicepteurs périphériques en ciblant les canaux sodiques nociceptifs NaV1.7 et NaV1.8. C’est un rappel important que l’analgésie peut impliquer des effets directs sur la machinerie de l’excitabilité, et pas seulement la signalisation GPCR. Un rapport de recherche de 2026 mis en avant par ScienceDaily a poussé la même idée translationnelle plus loin, en décrivant un composé dérivé du cannabis qui soulageait la douleur sans le high. Cela reste une preuve au stade de la recherche, pas une médecine établie, mais cela pointe vers une stratégie sérieuse de développement : séparer l’analgésie de l’intoxication centrale en exploitant des cibles non CB1, une restriction périphérique, ou les deux.
La chimie médicinale va dans cette direction. Le criblage structurel autour des récepteurs cannabinoid était déjà bien établi avec l’article de 2016 du Journal of Medicinal Chemistry « Library Docking for Cannabinoid-2 Receptor Ligands », et les efforts plus récents visent de plus en plus des mécanismes différenciés plutôt que de simples composés « un peu comme le THC mais plus faibles ». Un communiqué de MIRA Pharmaceuticals de 2025, qui doit être traité comme des données précliniques rapportées par l’entreprise et non comme une confirmation indépendante, décrivait MIRA-55 comme ayant un mécanisme différencié et une activité anxiolytique par rapport au THC. Le point important n’est pas que l’affirmation est prouvée. C’est que les développeurs de médicaments supposent désormais que la séparation des cibles importe.
Cela nous ramène à l’entourage effect. En tant qu’idée scientifique étroite, il est plausible que des combinaisons de cannabinoid, de terpènes et de métabolites modifient la pharmacocinétique ou produisent des effets additifs, opposés, ou parfois supra-additifs sur plusieurs cibles. En tant qu’explication globale du fait que n’importe quel produit de cannabis mixte serait supposément meilleur, il est généralement trop vague pour être testé et trop élastique pour être falsifié.
Le problème chimique est fondamental. Un mélange contenant THC, CBD, CBG, des cannabinoid acides, des produits oxydés, des terpènes résiduels et des métabolites variables n’est pas une seule intervention. C’est un ensemble de pièces en mouvement dont les effets dépendent des rapports de concentration et du timing. Une pharmacologie multi-cibles plausible existe. Une simplification non étayée existe aussi. Ce ne sont pas les mêmes choses.
Le meilleur standard est une preuve spécifique à la cible, liée à l’exposition. Quel composé, à quelle concentration, dans quel tissu, produisant quel effet mesurable ? Une fois ces questions posées, la mythologie s’efface et la véritable histoire des cannabinoid apparaît : non pas deux récepteurs, mais une carte pharmacologique dense.
La méthode compte : pourquoi la conception des essais façonne ce que nous pensons que font les cannabinoid
Les affirmations concernant les cibles des cannabinoid semblent souvent plus nettes que les données qui les sous-tendent. Un article dit que CBD « active TRPV1 », un autre dit qu’il s’agit d’un agoniste de 5-HT1A, un troisième appelle GPR55 un candidat récepteur cannabinoid, et une étude de docking propose une pose élégante dans CB2 ou dans la poche d’un GPCR orphelin. Ces déclarations peuvent être utiles sur le plan directionnel. Elles ne constituent pas toutes le même type de preuve.
Cette distinction importe parce que les cannabinoid sont des molécules grasses, aimant les membranes, avec la mauvaise habitude de paraître actives dans davantage d’endroits qu’ils ne comptent réellement in vivo. Le domaine l’a appris à ses dépens. Si un composé se partitionne dans les membranes, modifie les propriétés de la bicouche, s’accumule à l’intérieur des cellules, forme des métabolites actifs ou ne montre un effet qu’à 10 à 50 micromolaires, il peut générer des récits de cibles qui s’effondrent lorsque les conditions sont plus strictes. Pour la pharmacologie du cannabis, la méthode n’est pas un détail technique. Elle détermine quelles explications survivent.
Essais de liaison, essais fonctionnels et études de docking
Commençons par la question la plus ancienne et la plus nette : le composé se lie-t-il ? Dans un essai de liaison radioligand, des membranes ou des cellules intactes exprimant un récepteur sont incubées avec un ligand radioactif connu et des concentrations croissantes du composé test. Si la molécule test déplace le radioligand, les chercheurs estiment l’affinité, souvent exprimée en Ki. C’est utile. C’est aussi limité. La liaison indique qu’une molécule peut occuper un site dans les conditions de l’essai ; elle ne dit pas ce qui se passe ensuite.
C’est pourquoi les essais fonctionnels comptent davantage que la simple liaison lorsque l’on formule des affirmations sur la signalisation. Pour les canaux TRP tels que TRPV1, TRPA1 ou TRPM8, les chercheurs utilisent souvent des essais de flux calcique avec des colorants fluorescents. Si l’ouverture du canal laisse entrer le calcium, la fluorescence augmente. L’avantage est évident : ces essais se prêtent à l’échelle et peuvent comparer rapidement de nombreux composés. Le problème est tout aussi évident si l’on connaît les cannabinoid. Certains sont fluorescents, certains perturbent les membranes, certains libèrent le calcium indirectement à partir des réserves intracellulaires, et certains déclenchent une désensibilisation du canal après activation initiale. Un seul pic dans une courbe calcique peut masquer plusieurs mécanismes.
Le patch-clamp est plus lent mais beaucoup plus informatif. Il enregistre directement le courant ionique. Pour les canaux ioniques, en particulier les canaux sodiques tels que NaV1.7 et NaV1.8, le patch-clamp peut montrer si le médicament modifie l’activation, l’inactivation, la probabilité d’ouverture ou la densité de courant. C’est pourquoi le rapport 2025 de l’Université hébraïque sur THC agissant sur les nocicepteurs périphériques via NaV1.7 et NaV1.8 est méthodologiquement important : l’électrophysiologie directe peut distinguer un véritable effet sur un canal d’un signal cellulaire vague. Si un cannabinoid réduit le courant sodique dans les nocicepteurs à des concentrations atteintes dans les tissus périphériques, cela a plus de poids mécanistique pour l’analgésie qu’une autre simple étiquette « interaction récepteur ».
Les GPCR et les récepteurs nucléaires exigent d’autres lectures. Pour les GPCR, les chercheurs peuvent mesurer l’AMPc, le recrutement de beta-arrestine, la liaison GTPγS, la phosphorylation d’ERK ou des signaux calciques dans des lignées cellulaires ingénierées. Ces lectures ne sont pas interchangeables. Un cannabinoid peut apparaître comme agoniste dans une voie, agoniste partiel dans une autre, et antagoniste dans une troisième. Ce n’est pas de la négligence ; c’est du biais de signalisation. Pour PPAR-gamma, l’approche commune est un essai rapporteur dans lequel un promoteur sensible à PPAR commande la luciférase. La lumière augmente, et le composé est appelé activateur de PPAR. Mais les essais rapporteurs sont éloignés de plusieurs étapes de l’engagement direct de la cible. Un cannabinoid lipophile peut modifier la transcription de manière indirecte par le stress cellulaire, le métabolisme ou des changements dans les médiateurs lipidiques endogènes.
Les lectures d’expression génique sont encore plus en aval. Si un article montre que CBD modifie les transcrits inflammatoires et que l’effet est réduit par un antagoniste de PPAR, cela est suggestif, pas définitif. L’antagoniste peut être imparfait. Les cellules peuvent produire des métabolites. Le cannabinoid peut modifier le tonus adénosinergique, la gestion du calcium, l’état redox ou l’ordre membranaire. Le mécanisme par soustraction est fragile.
Puis vient le docking. Le docking demande si une petite molécule peut entrer dans une poche de liaison modélisée ou déterminée expérimentalement et obtenir un score favorable. Utilisé correctement, c’est un triage de chimie médicinale. Utilisé mal, il devient une certitude décorative. L’article de 2016 de l’ACS Journal of Medicinal Chemistry, « Library Docking for Cannabinoid-2 Receptor Ligands », est une bonne porte d’entrée parce qu’il montre la logique à son meilleur : le criblage fondé sur la structure peut aider à prioriser des squelettes, identifier des contacts ligand-récepteur plausibles et guider la synthèse autour de la sélectivité pour CB2. C’est à cela que sert le docking. Il génère des hypothèses. Il ne prouve pas qu’un phytocannabinoid engage une cible dans un tissu vivant, encore moins que l’interaction explique l’analgésie, l’anxiété ou l’inflammation.
Différences entre espèces, métabolites et effets membranaires
Même des signaux in vitro solides peuvent échouer hors de la boîte d’essai parce que la pharmacologie cannabinoid dépend fortement du contexte. Les récepteurs humains et murins ne sont pas toujours fonctionnellement identiques. Un composé actif sur les lectures TRPA1 murines ou 5-HT1A liées au rat peut voir sa puissance ou son efficacité changer avec l’orthologue humain. La même mise en garde vaut pour les variantes d’épissage, la réserve réceptrice et le contexte cellulaire. Une lignée cellulaire à surexpression massive du récepteur peut faire paraître important un ligand faible.
Le métabolisme ajoute une autre couche. De nombreux cannabinoid ne restent pas longtemps sous leur forme parentale. THC devient 11-hydroxy-THC ; d’autres structures forment des métabolites oxydés ou conjugués dont le profil cible peut différer fortement. Les régulateurs prêtent davantage attention à ce problème dans des débats voisins sur les politiques en matière de drogues. En 2025, HHS a déclaré que « 7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety », soulignant une leçon plus large : les intoxicants renforcés ou métaboliquement avantagés peuvent se comporter très différemment du composé parent. Le cannabis pose des problèmes parallèles avec les cannabinoid semi-synthétiques, les nouveaux isomères et les formulations conçues pour modifier l’exposition. Si vous n’essayez que la molécule parentale, vous pouvez manquer l’espèce qui détermine réellement les effets in vivo.
Les membranes sont le plus grand confondant silencieux. Les cannabinoid sont suffisamment lipophiles pour s’accumuler dans les bicouches et les compartiments intracellulaires. Cela signifie que la concentration nominale dans le bain est souvent un mauvais substitut de la concentration sur la cible. Une application à 10 micromolaires dans un essai cellulaire peut créer une charge membranaire locale très élevée, susceptible de modifier le gating des canaux ou le comportement des récepteurs de manière non spécifique. Elle peut aussi produire de faux négatifs si la concentration aqueuse libre est bien plus faible qu’attendu parce que le composé adhère au plastique, aux protéines sériques ou à la membrane elle-même.
C’est l’une des raisons pour lesquelles les affirmations à haute concentration méritent le scepticisme. Si un cannabinoid n’affecte une cible qu’au-delà de 20 ou 30 micromolaires, la première question doit être de savoir si cela reflète une interaction physiologiquement pertinente ou un artefact lié aux membranes. Les canaux TRP sont particulièrement vulnérables à la surinterprétation ici. Ce sont de véritables cibles sensibles aux cannabinoid dans certains cas, mais les effets peuvent être biphasés, se désensibiliser rapidement et être très sensibles à la concentration. Une brève poussée calcique à forte concentration micromolaire ne signifie pas automatiquement un mécanisme thérapeutiquement pertinent.
Des articles de docking ACS à la pharmacologie réelle
La chimie médicinale prospère grâce à la simplification, mais la biologie punit la simplification excessive. L’article de docking sur CB2 de l’ACS illustre la version utile de la simplification : partir d’une structure de récepteur, cribler des bibliothèques, prioriser des candidats, synthétiser des analogues, les tester dans des essais de liaison et fonctionnels, puis apprendre à partir des relations structure-activité. Cette séquence peut conduire à de vrais médicaments. Elle peut aussi révéler que la pose in silico la plus séduisante appartient à un composé à la perméabilité médiocre, au métabolisme instable, à la signalisation biaisée ou sans activité significative dans des cellules natives.
La distance entre la pose dockée et la pharmacologie est précisément l’endroit où de nombreuses affirmations sur les cannabinoid échouent. Une image de docking de CBD ou CBG dans TRPV1, 5-HT1A, GPR55 ou PPAR-gamma ne prouve pas que le composé détermine la physiologie pertinente chez l’animal ou chez l’humain. Une vraie pharmacologie demande une convergence. Idéalement, cela signifie un engagement direct de la cible à des concentrations plausibles, des effets fonctionnels dans des systèmes natifs, des preuves de perte de fonction via antagonistes ou knock-outs, un soutien pharmacocinétique, et un lien avec le comportement ou l’issue clinique.
Lorsque ces niveaux concordent, l’histoire devient rapidement plus solide. La poussée actuelle de recherche visant à séparer l’analgésie de l’intoxication dépend de cette logique. Le rapport ScienceDaily de 2026 décrivant « a cannabis compound that relieves pain without the high » est intéressant précisément parce qu’il dépasse l’équivalence grossière au THC et pointe vers des mécanismes sélectifs ou périphériquement restreints. Il en va de même pour les travaux sur les canaux sodiques concernant THC. Il en va de même pour les efforts de construction de ligands sélectifs par docking et conception guidée par la structure. Le schéma est cohérent même lorsque certaines affirmations individuelles doivent être nuancées.
Ce que la pipeline ne ressemble probablement pas à court terme, en revanche, est un succès clinique massif. La promiscuité des cibles, la complexité des métabolites, les différences d’espèce et les effets de formulation continuent à briser les récits simples. Mais le domaine n’est plus bloqué à la question de savoir si les cannabinoid sont « vraiment » à propos de CB1 et CB2. Les développeurs de médicaments ont déjà répondu à cette question avec leurs budgets de chimie. Ils conçoivent pour l’espace au-delà de ces récepteurs, car c’est là que se trouve aujourd’hui la meilleure chance de séparer le bénéfice de la toxicité.
Niveaux de preuve : de la boîte de culture au clinic
La littérature sur les cibles hors CB1/CB2 est riche parce que les cannabinoid sont chimiquement promiscues. Une seule molécule peut toucher TRPV1, une signalisation liée à 5-HT1A, PPAR-gamma, GPR55, le tonus adénosinergique et les canaux sodiques voltage-dépendants selon la concentration, le tissu et le profil métabolique. Cela fait de bons articles mécanistiques. Cela ne produit pas automatiquement une médecine prouvée. S’il y a une règle qui maintient ce domaine honnête, elle est simple : chaque marche supplémentaire de l’échelle des preuves élimine une grande part des affirmations qui paraissaient convaincantes en dessous.
Ce que les preuves précliniques peuvent et ne peuvent pas démontrer
Au bas de l’échelle se trouvent les essais de liaison, les enregistrements de canaux, les systèmes rapporteurs et les cultures cellulaires. Ces méthodes sont indispensables. C’est grâce à elles que les chercheurs ont appris que la pharmacologie cannabinoid va bien au-delà des récepteurs classiques, et c’est pourquoi les explications du cannabis réduites aux récepteurs paraissent aujourd’hui datées. Le prix Nobel de physiologie ou médecine 2021 a reconnu David Julius et Ardem Patapoutian « for their discoveries of receptors for temperature and touch », rappelant que la biologie TRP n’est pas un trivia périphérique ; elle se situe près du centre de la science moderne de la douleur. Lorsque les cannabinoid activent ou désensibilisent TRPV1, TRPA1 ou des canaux apparentés dans une boîte, ce résultat compte.
Mais une boîte ne peut pas dire si le même engagement de cible se produit à des doses humaines tolérées. De nombreux effets in vitro des cannabinoid n’apparaissent qu’à des concentrations micromolaires. Les niveaux plasmatiques après un dosage réel peuvent être plus faibles, plus transitoires ou déplacés vers des métabolites aux pharmacologies différentes. CBD en est l’exemple classique. Il montre à répétition des actions difficiles à expliquer uniquement par CB1 ou CB2, ce qui explique pourquoi les voies TRPV1, 5-HT1A, PPAR-gamma, GPR55 et l’adénosine réapparaissent sans cesse dans la littérature. Pourtant, le fait que CBD module une cible dans des cellules en culture ne prouve pas que ce mécanisme détermine une issue clinique dans l’épilepsie, l’anxiété, la douleur ou l’inflammation.
Les modèles animaux constituent une étape plus proche de la médecine, sans être encore la médecine. Des études chez le rongeur peuvent montrer qu’un cannabinoid réduit l’allodynie, supprime des marqueurs inflammatoires ou modifie le comportement de type anxieux. Elles peuvent même étayer des récits spécifiques de cible avec des antagonistes, des knock-outs ou une restriction périphérique. Le rapport récent de l’Université hébraïque en est un bon exemple : des chercheurs ont rapporté que THC inhibe les nocicepteurs périphériques en ciblant les canaux sodiques nociceptifs NaV1.7 et NaV1.8. Cette découverte va à l’encontre de l’hypothèse paresseuse selon laquelle l’analgésie liée au THC doit être une histoire CB1 plus intoxication. Si l’effet se confirme, il suggère une action pertinente pour la douleur au niveau même de l’excitabilité périphérique.
Néanmoins, même des travaux solides chez l’animal ne peuvent prouver que le blocage de NaV1.7/NaV1.8 par un cannabinoid deviendra un analgésique humain sûr et efficace. Les différences d’espèce comptent. Le dosage compte. La voie d’administration compte. Les lectures comportementales chez la souris peuvent être trompeuses. Un signal douloureux atténué dans une préparation nerveuse ou un test au formol peut ne pas se traduire par un soulagement de la neuropathie, de l’arthrose ou de la douleur post-opératoire chez l’humain. La même prudence s’applique aux annonces des entreprises. En 2025, MIRA Pharmaceuticals a déclaré que son candidat MIRA-55 présentait un « differentiated mechanism of action » et une « anxiolytic activity relative to THC » dans des données précliniques. C’est légitime comme preuve au stade de la recherche. Ce n’est pas une revendication thérapeutique, et cela ne doit pas être traité comme telle.
La même prudence translationnelle s’applique aux titres séduisants. ScienceDaily a mis en avant en 2026 des travaux sur « a cannabis compound that relieves pain without the high ». Peut-être. C’est une direction utile pour la conception de médicaments, en particulier si des cibles périphériques ou des mécanismes non CB1 peuvent dissocier l’analgésie de l’intoxication centrale. Mais « sans le high » dans un rapport préclinique est une hypothèse à l’étude, pas un fait établi concernant une thérapie humaine.
Médicament cannabinoid approuvé et étroitesse des indications actuelles
Passons maintenant au sommet de l’échelle : les preuves de médicament approuvé. Ici, le domaine devient beaucoup plus étroit. L’exemple américain le plus clair est la solution orale de cannabidiol. La notice approuvée par la FDA indique qu’elle est indiquée « for the treatment of seizures associated with Lennox-Gastaut syndrome, Dravet syndrome, or tuberous sclerosis complex in patients 1 year of age and older ». Cette phrase est plus informative que des dizaines d’allégations bien-être vagues, car elle nomme la forme du médicament, le résultat, les maladies et la tranche d’âge.
Remarquez ce que la notice ne dit pas. Elle ne dit pas que CBD est approuvé pour la douleur, l’anxiété généralisée, l’insomnie, la maladie inflammatoire de l’intestin, la neuroprotection ou un vague « équilibre endocannabinoïde ». Elle ne valide pas non plus tous les mécanismes proposés dans la littérature préclinique. L’approbation nous apprend qu’un produit précis à base de cannabidiol a démontré une efficacité et une sécurité acceptables pour des syndromes épileptiques spécifiques dans des études contrôlées. Elle ne règle pas la question de savoir si TRPV1, GPR55, 5-HT1A, PPAR-gamma, les effets sur le calcium intracellulaire ou les actions métaboliques sont le mécanisme clinique dominant. Le mécanisme peut rester partiellement non résolu même lorsque l’efficacité est réelle.
Cet écart entre indication approuvée et folklore mécanistique est courant en pharmacologie. Beaucoup de médicaments ont été efficaces chez les patients avant que leur carte complète des cibles soit comprise. Les cannabinoid ne font pas exception. Ce qui est inhabituel, c’est la fréquence à laquelle des affirmations larges sont reconstruites à partir de résultats dispersés sur des récepteurs, puis présentées comme si elles avaient déjà passé le test clinique.
L’étroitesse des indications actuelles importe aussi pour les discussions de santé publique. Les régulateurs distinguent de plus en plus les cannabinoid végétaux familiers des intoxicants altérés, renforcés ou semi-synthétiques avec des profils de risque différents. En 2025, HHS a déclaré que « 7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety » en soutenant une action de classement sur des produits 7-OH renforcés. Cette déclaration concernait des produits liés au kratom, et non le cannabis, mais la leçon politique se transpose proprement : dès que des fabricants commencent à enrichir, modifier ou synthétiser des molécules psychoactives puissantes, le raccourci fondé sur l’origine végétale devient peu fiable. La pharmacologie au niveau des cibles commence alors à compter beaucoup.
Pourquoi les récits mécanistiques dépassent souvent les données cliniques
Ils les dépassent parce que les récits mécanistiques sont rapides, vivants et publiables. La preuve clinique est lente et coûteuse. Un article cellulaire peut montrer qu’un cannabinoid active TRPA1, antagonise GPR55 ou modifie la signalisation 5-HT1A en quelques semaines de travail. Un essai randomisé convaincant sur la douleur chronique peut prendre des années et échouer malgré tout parce que la taille d’effet est faible, les effets indésirables limitent la dose, ou la cible préclinique n’a jamais été significativement engagée chez les patients.
La chimie cannabinoid invite aussi à la surinterprétation. Des composés structurellement proches peuvent avoir des profils de cibles très différents, et le métabolisme peut remodeler le tableau après administration. La voie d’administration change encore l’histoire. L’administration orale produit des métabolites de premier passage ; l’inhalation modifie la cinétique ; les formulations topiques ou périphériques peuvent favoriser des cibles locales au détriment du système central. Même la catégorie juridique « hemp » dit peu de choses sur le plan pharmacologique. Le Farm Bill de 2018 a fixé la limite à un delta-9 THC « not more than 0.3 percent on a dry weight basis », un seuil statutaire, pas biologique.
La bonne lecture des preuves hors CB1/CB2 n’est donc ni un rejet ni un engouement. La littérature préclinique montre réellement que les cannabinoid agissent sur autre chose que CB1 et CB2. Pour la douleur surtout, les canaux TRP et les canaux sodiques méritent une attention sérieuse. Pour CBD, les cibles non classiques ne sont pas des notes de bas de page facultatives ; elles sont probablement centrales pour expliquer son profil. Mais la plausibilité n’est pas l’efficacité, l’engagement de la cible n’est pas le bénéfice pour le patient, et l’approbation d’un médicament cannabinoid pour un petit ensemble de syndromes épileptiques ne ratifie pas le nuage beaucoup plus vaste d’affirmations fondées sur des schémas récepteurs, le comportement murin et des résultats de boîtes de culture.
Sécurité, réglementation et pourquoi la pharmacologie hors cible compte en santé publique
Les problèmes de santé publique ne s’alignent pas proprement sur les schémas récepteurs. Un cannabinoid peut être d’origine végétale, issu du hemp, semi-synthétique ou entièrement synthétique et finir malgré tout par produire des risques mal prédits par son étiquette, son origine ou sa catégorie juridique. C’est le sens pratique de la pharmacologie hors cible. Dès qu’une molécule touche les canaux TRP, les récepteurs de la sérotonine, les PPAR, les systèmes de signalisation de type GPR55, les canaux sodiques et d’autres cibles non CB1/CB2, le profil de sécurité peut changer d’une manière pertinente pour la surveillance des intoxications, les standards de produit, la politique de conduite sous influence et le risque de dépendance.
L’erreur n’est pas seulement scientifique. Elle est réglementaire. Les politiques concernant le cannabis ont souvent traité « cannabinoid » comme si cela constituait déjà une explication.
Intoxicants renforcés et leçon politique de 7-OH
L’avertissement actuel le plus clair ne provient même pas d’un constituant classique du cannabis. En 2025, le U.S. Department of Health and Human Services a déclaré que « 7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety » en soutenant une action de la DEA contre des produits 7-OH renforcés. Cette phrase compte parce qu’elle montre des autorités sanitaires fédérales traçant une ligne entre une exposition botanique familière et des intoxicants concentrés ou chimiquement manipulés pouvant se comporter très différemment dans les populations réelles.
La leçon politique se transpose directement aux cannabinoid. Une catégorie large telle que « dérivé du hemp », « d’origine végétale » ou même « cannabinoid-like » n’indique presque rien aux régulateurs sur les cibles qu’un composé engage aux concentrations pertinentes en usage. Elle n’indique presque rien non plus aux consommateurs sur la puissance, le délai d’action, la durée, le risque d’interaction ou le potentiel d’abus.
Les produits 7-OH renforcés sont devenus un problème de santé publique parce que la chimie a modifié l’exposition. Lorsqu’un marché passe d’une occurrence naturelle à l’état de trace à un ingrédient actif concentré, la pharmacologie cesse d’être une question de trivia et devient le problème central de sécurité. Le même schéma est déjà apparu autour des cannabinoid semi-synthétiques et structurellement modifiés vendus sous l’égide de la légalité hemp après que le Farm Bill de 2018 a défini le hemp par une concentration de delta-9 THC de « not more than 0.3 percent on a dry weight basis ». Ce seuil en poids sec est une définition de culture, pas un standard pharmacologique. Il ne filtre ni l’activité TRP, ni le blocage des canaux sodiques, ni la signalisation 5-HT1A, ni les effets de GPR55, ni les métabolites plus persistants et à pénétration centrale différente.
C’est pourquoi les effets hors cible ne sont pas un détail obscur. Ils constituent l’une des voies par lesquelles des produits commercialisés comme adjacents au cannabis peuvent générer des dommages inattendus. Un composé présenté au public comme « THC-like but legal » peut différer par son efficacité intrinsèque sur CB1, mais il peut aussi différer de manières beaucoup moins visibles : stimulation cardiovasculaire plus forte, potentiel proconvulsivant ou anxiogène accru, dysphorie plus sévère, sédation inhabituelle, ou profil toxique façonné par le métabolisme plutôt que par le seul médicament parent. Ces possibilités ne sont pas abstraites ; elles expliquent précisément pourquoi les agences sanitaires réagissent différemment aux intoxicants renforcés qu’aux grandes catégories végétales.
Une réponse politique saine commence par des questions au niveau des cibles. Quels récepteurs et canaux sont engagés ? À quelles concentrations ? Dans quels tissus ? Quels sont les principaux métabolites ? Existe-t-il des preuves d’inhibition des canaux sodiques susceptible de modifier la nociception ou la conduction cardiaque ? L’activation de TRPV1 est-elle susceptible de désensibiliser la signalisation douloureuse, ou d’irriter et d’aggraver les symptômes à faibles expositions ou expositions transitoires ? Ce sont des questions plus difficiles que « est-ce un cannabinoid », mais ce sont les bonnes.
Les limites de la pensée en équivalence THC
L’équivalence au THC est séduisante parce qu’elle simplifie le droit, la fiscalité, l’étiquetage et les discussions sur l’altération. Elle est aussi souvent fausse. Deux composés peuvent produire un certain degré d’intoxication tout en différant fortement en matière de risque anxieux, de potentiel psychotomimétique, d’effet analgésique, de réponse cardiaque, de contrôle des vomissements, de développement de tolérance et de charge de sevrage, parce qu’ils ne partagent pas la même carte élargie des cibles.
Même THC lui-même n’est pas pharmacologiquement épuisé par CB1 et CB2. En 2025, des chercheurs de l’Université hébraïque ont rapporté que THC inhibe les nocicepteurs périphériques en ciblant les canaux sodiques nociceptifs NaV1.7 et NaV1.8. Ce résultat va à l’encontre des récits simplistes du type « THC agit ici, CBD agit là-bas ». Si un cannabinoid psychoactif canonique peut affecter directement des canaux sodiques voltage-dépendants pertinents pour la douleur, alors la sécurité et l’efficacité ne peuvent pas être déduites du seul branding cannabinoid. La dose, la voie d’administration, la distribution et l’exposition tissulaire deviennent déterminantes.
Le même point apparaît à l’envers avec CBD. Sa solution orale approuvée est indiquée par la FDA pour les crises associées au syndrome de Lennox-Gastaut, au syndrome de Dravet ou au complexe de sclérose tubéreuse chez les patients âgés d’un an et plus, un fait clinique qui n’a jamais été bien expliqué par l’agonisme CB1 parce que CBD n’est pas un intoxiquant CB1 classique. Son profil a depuis longtemps poussé les chercheurs vers d’autres mécanismes, notamment TRPV1, 5-HT1A, la signalisation liée à l’adénosine et PPAR-gamma. Tous ces mécanismes ne sont pas également établis chez l’humain, mais ensemble ils rendent une chose inévitable : les effets cannabinoid reposent souvent sur un réseau, et non sur un seul interrupteur récepteur.
Cette perspective en réseau explique aussi pourquoi l’intoxication est un mauvais indicateur principal de sécurité publique. Un produit peut être moins intoxicant que THC tout en produisant des interactions médicamenteuses préoccupantes, des effets enzymatiques hépatiques, une contrainte cardiovasculaire, des réactions paniques ou de la sédation. Il peut aussi être plus intoxicant sans être plus prévisible. Le public entend souvent « plus faible que THC » ou « plus fort que THC » comme si cela réglait la question. Ce n’est pas le cas. Il s’agit généralement d’une seule phénotype saillante, pas d’un profil toxicologique complet.
Le pipeline de recherche s’éloigne déjà de l’équivalence THC. Un communiqué Nasdaq de MIRA Pharmaceuticals en 2025 décrivait des données précliniques pour MIRA-55, affirmant un « differentiated mechanism of action » et une activité anxiolytique relative au THC. Les communiqués d’entreprise sont des preuves faibles par rapport aux données cliniques évaluées par les pairs, de sorte que l’affirmation doit être traitée avec prudence. Néanmoins, la direction est réelle : la chimie médicinale cherche à séparer les effets souhaités de l’intoxication centrale en modifiant l’engagement des cibles. La même logique translationnelle apparaît dans un rapport ScienceDaily de 2026 décrivant des travaux sur « a cannabis compound that relieves pain without the high ». Les résultats au stade de la recherche ne sont pas des preuves cliniques, mais ils renforcent un point réglementaire central. Si des effets bénéfiques peuvent être séparés de l’intoxication, alors les dommages peuvent aussi l’être. Un produit à faible high n’est pas automatiquement un produit à faible risque.
Pourquoi les cannabinoid nouveaux exigent un examen au niveau des cibles
Les cannabinoid nouveaux méritent plus de surveillance, et non moins, précisément parce qu’ils sont souvent introduits sur le marché avant que leur pharmacologie chez l’humain soit cartographiée. L’ancien raccourci consistait à demander si une molécule se liait à CB1 ou CB2. La meilleure question est de savoir ce qu’elle fait d’autre, et si ces actions deviennent pertinentes à des doses réelles après inhalation, ingestion orale ou métabolisme.
Pour la douleur, les canaux TRP et sodiques sont des exemples évidents. David Julius et Ardem Patapoutian ont reçu le prix Nobel de physiologie ou médecine 2021 pour leurs découvertes sur les récepteurs de la température et du toucher, rappelant que la biologie somatosensorielle est fondée sur des cibles que les cannabinoid peuvent influencer en dehors des récepteurs cannabinoid classiques. L’activation de TRPV1 peut contribuer à l’analgésie par désensibilisation, mais elle peut aussi produire une irritation et dépend fortement de la concentration. Un régulateur qui ne chercherait que l’activité CB1 manquerait tout cet axe risque-bénéfice.
Pour la sécurité psychiatrique, la signalisation sérotoninergique compte. CBD a été à plusieurs reprises présenté comme un candidat anxiolytique lié à 5-HT1A, mais l’implication de la sérotonine peut être indirecte et dépendante du contexte. Cette incertitude n’est pas une raison d’ignorer la cible ; c’est une raison de l’étudier soigneusement avant de normaliser les produits comme inoffensifs. Il en va de même pour GPR55, parfois présenté comme candidat « CB3 » malgré la controverse persistante. Une cible contestée reste une cible capable de façonner la signalisation calcique, l’excitabilité et les réponses inflammatoires.
Pour les effets métaboliques et inflammatoires, des cibles intracellulaires comme PPAR-gamma compliquent encore le tableau. La signalisation par récepteur nucléaire ne ressemble pas à un effet d’intoxication rapide, mais elle peut compter pour l’exposition chronique, la distribution adipeuse, les changements transcriptionnels et les interactions avec d’autres états pathologiques. Les messages de santé publique construits autour de la question de savoir si quelque chose « vous fait planer » manquent ces risques plus lents et plus discrets.
C’est ici que la réglementation, la toxicologie et la communication au consommateur ont besoin d’une littératie des cibles moléculaires. Tous les signaux in vitro ne compteront pas cliniquement. Les différences entre espèces sont réelles. Les métabolites peuvent dominer. Mais les agences devraient exiger des panels de cibles, la caractérisation des métabolites, des données concentration-réponse et des études humaines pertinentes pour l’altération avant d’assumer qu’un nouveau cannabinoid peut être gouverné comme un autre. L’article de 2016 du Journal of Medicinal Chemistry « Library Docking for Cannabinoid-2 Receptor Ligands » reflète la manière dont la découverte de médicaments fonctionne désormais : conception fondée sur la structure, ingénierie de la sélectivité et attention explicite aux différences au niveau des récepteurs. La réglementation devrait cesser de prétendre que le marché est plus simple que la chimie médicinale ne le sait déjà.
La santé publique ne peut pas se permettre le réductionnisme récepteur. « Cannabinoid » est une étiquette de départ. Ce n’est pas une conclusion de sécurité.
Découverte de médicaments : concevoir des cannabinoid et des molécules inspirées des cannabinoid pour des cibles autres que CB1/CB2
La découverte de médicaments autour des cannabinoid est allée bien au-delà de l’ancienne question de savoir si une molécule est « active sur CB1 » ou « active sur CB2 ». Cette simplification a toujours été fragile, car de nombreux composés apparentés aux cannabinoid sont pharmacologiquement désordonnés : ils ciblent des canaux ioniques, des GPCR autres que la paire canonique, des récepteurs nucléaires intracellulaires, des processus de transport et des enzymes métabolisantes, souvent avec des effets différents selon la concentration, le tissu et la voie d’administration. Pour les chimistes médicinaux, ce désordre n’est pas seulement un problème. C’est aussi une opportunité.
L’objectif de conception central est assez clair : conserver les effets analgésiques, anti-inflammatoires ou anxiolytiques, tout en réduisant les inconvénients liés à une forte activation centrale de CB1. Ces inconvénients ne sont pas abstraits. La sédation, l’altération cognitive, l’intoxication, le potentiel d’abus et les effets psychiatriques limitant la dose sont précisément la raison pour laquelle « THC-like » est souvent un mauvais profil dans un programme de développement, même lorsque THC lui-même montre une pharmacologie utile. Le climat réglementaire actuel renforce ce point. En 2025, HHS a soutenu une action de classement en déclarant que « 7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety », rappelant que des intoxicants modifiés ou renforcés ne peuvent pas être supposés se comporter comme des composés végétaux familiers simplement parce qu’ils se trouvent à proximité sur une étagère marketing. La pharmacologie au niveau des cibles importe.
Restriction périphérique, sélectivité fonctionnelle et biais de signalisation
Une voie pour contourner les effets indésirables centraux est simple mais efficace : empêcher le médicament d’entrer dans le cerveau. La restriction périphérique peut être conçue en augmentant la surface polaire, en accroissant la capacité de liaison aux hydrogènes, en ajustant le pKa ou en faisant de la molécule un substrat des transporteurs d’efflux de la barrière hémato-encéphalique. L’idée n’est pas propre à la science cannabinoid, mais elle s’y adapte particulièrement bien parce que la signalisation de la douleur commence souvent dans les nocicepteurs périphériques, les tissus inflammés et les ganglions rachidiens dorsaux.
C’est là que les cibles hors CB1/CB2 deviennent particulièrement attrayantes. Un rapport 2025 de l’Université hébraïque a soutenu que THC inhibe les nocicepteurs périphériques en ciblant les canaux sodiques NaV1.7 et NaV1.8, deux moteurs majeurs de l’excitabilité nociceptive. Si ce mécanisme se confirme dans différents systèmes, il aura beaucoup d’importance. NaV1.7 est depuis longtemps considéré comme une cible de choix pour la douleur parce que les mutations perte de fonction dans SCN9A peuvent provoquer une insensibilité congénitale profonde à la douleur. Un squelette cannabinoid qui conserve la modulation des canaux sodiques en périphérie tout en minimisant la signalisation centrale de CB1 ne serait pas simplement un « THC moins intoxicant ». Ce serait un type différent d’analgésique.
Les canaux TRP ouvrent une ouverture similaire. La biologie sensorielle plus large a été reconnue au plus haut niveau lorsque le prix Nobel de physiologie ou médecine 2021 a été décerné à David Julius et Ardem Patapoutian pour leurs découvertes de récepteurs de la température et du toucher. TRPV1, TRPA1 et des canaux apparentés sont profondément liés à la nociception et à la signalisation inflammatoire, et plusieurs phytocannabinoid interagissent avec eux de manière sensible à la concentration et parfois paradoxale : une activation initiale peut être suivie d’une désensibilisation, cette dernière pouvant contribuer à l’analgésie. Cela rend la chimie médicinale plus difficile, pas plus facile. Mais cela signifie aussi qu’un composé n’a pas besoin d’être un ligand orthostérique propre, de type tout-ou-rien, des récepteurs cannabinoid pour avoir une valeur thérapeutique.
La sélectivité fonctionnelle ajoute une couche supplémentaire. Même au niveau de CB1 ou CB2, des ligands peuvent favoriser une sortie de signalisation plutôt qu’une autre, en modifiant l’équilibre entre les voies de la protéine G, le recrutement de beta-arrestine, l’internalisation du récepteur et les programmes transcriptionnels en aval. En termes simples, deux molécules peuvent toutes deux « se lier à CB1 » et pourtant se comporter de manière très différente dans un tissu vivant. L’article de 2016 du Journal of Medicinal Chemistry « Library Docking for Cannabinoid-2 Receptor Ligands » reflète à quel point la stratégie de conception a évolué vers un ajustement fondé sur la structure plutôt qu’une simple étiquette récepteur. La même logique s’étend désormais au-delà des récepteurs cannabinoid : les chimistes veulent des squelettes dont la forme, la lipophilie et les contraintes conformationnelles les orientent vers un mécanisme pertinent pour la douleur ou l’anxiété tout en évitant le lourd bagage de la signalisation centrale.
CBD constitue la preuve permanente qu’un médicament apparenté aux cannabinoid peut être important cliniquement sans s’expliquer par un agonisme CB1/CB2. L’étiquetage FDA de la solution orale de cannabidiol, mis à jour en 2024, couvre les crises associées au syndrome de Lennox-Gastaut, au syndrome de Dravet et au complexe de sclérose tubéreuse chez les patients âgés d’un an et plus. Quelle que soit la combinaison de TRPV1, 5-HT1A, GPR55, signaux adénosinergiques, effets intracellulaires et effets de réseau qui explique finalement cette efficacité, ce n’est pas une simple histoire CB1. Les chercheurs en découverte de médicaments l’ont remarqué.
MIRA-55 et la recherche de mécanismes différenciés
MIRA-55 est une étude de cas utile non pas parce qu’elle tranche la question, mais parce qu’elle montre comment les entreprises présentent désormais les programmes cannabinoid. Dans un communiqué relayé par Nasdaq en 2025, MIRA Pharmaceuticals a indiqué que son candidat présentait un « differentiated mechanism of action » et une « anxiolytic activity relative to THC » dans des travaux précliniques. Cette formulation dit presque tout de la signalisation actuelle auprès des investisseurs et des régulateurs. Être inspiré des cannabinoid ne suffit plus. Une entreprise veut prendre ses distances avec la simple imitation du THC, surtout pour des indications comme l’anxiété où des effets centraux indésirables peuvent effacer le bénéfice.
La prudence reste néanmoins obligatoire. Un « differentiated mechanism » dans un communiqué d’entreprise est une affirmation, pas une conclusion. Il peut signifier un engagement récepteur modifié, une distribution tissulaire différente, des métabolites distincts, un agonisme partiel, un biais fonctionnel, des effets hors cible sur les canaux ioniques, ou simplement un profil comportemental différent dans un seul test animal. Sans panels pharmacologiques complets, courbes concentration-réponse, identification des métabolites, données d’occupation réceptrice et réplication à l’aveugle, l’expression relève davantage de l’hypothèse que du résultat.
Cela dit, la stratégie derrière cette affirmation est crédible. Si un composé peut réduire des comportements de type anxieux tout en diminuant l’intoxication, les troubles mnésiques ou la suppression locomotrice par rapport au THC, la chimie médicinale a probablement modifié un ou plusieurs des éléments suivants : la pénétration cérébrale, l’efficacité intrinsèque sur CB1, l’engagement de cibles non CB telles que 5-HT1A ou les canaux TRP, ou la conversion métabolique en espèces actives ayant un profil cible différent. Ce sont précisément les axes sur lesquels les programmes cannabinoid modernes se concurrencent.
MIRA-55 illustre aussi le problème de déconvolution des cibles. Les molécules de type cannabinoid sont souvent « sales » au sens pharmacologique. Ce n’est pas un jugement moral ; c’est un avertissement mécanistique. Si un signal anxiolytique préclinique apparaît, on ne peut pas supposer qu’un seul récepteur l’explique. La signalisation sérotoninergique peut être directe ou indirecte. Les effets de PPAR-gamma peuvent exiger une accumulation intracellulaire ou des métabolites. GPR55 peut paraître important dans un essai et marginal dans un autre. Un hit in vitro à 10 micromolaires peut être sans importance si les concentrations libres dans le cerveau n’approchent jamais ce niveau in vivo.
À quoi ressemble vraiment un pipeline cannabinoid réaliste
Un pipeline réaliste est plus étroit et plus discipliné que ne le suggère la rhétorique publique. Ce n’est pas un défilé de « non-psychoactive cannabis compounds » courant vers l’approbation. C’est un filtre.
En amont se trouvent la modification et le triage des squelettes : cannabinoid classiques, cannabinoid non classiques, lipides inspirés du système endocannabinoïde et chimiotypes non apparentés qui imitent une caractéristique utile de la pharmacologie cannabinoid sans hériter de tout le paquet. Les chimistes modifient la longueur de la chaîne latérale, les contraintes de cycle, la stéréochimie, la position des hétéroatomes et les points faibles métaboliques, puis testent non seulement CB1 et CB2, mais aussi TRPV1, TRPA1, les canaux NaV, certains GPCR orphelins et des essais pertinents pour la sérotonine. Les composés prometteurs passent ensuite par l’ADME, l’analyse des concentrations non liées et les mesures cerveau/plasma. Beaucoup échouent là.
Les programmes qui survivent se répartissent généralement en quelques catégories crédibles. L’une d’elles est celle des analgésiques à restriction périphérique, où le rêve est un soulagement de la douleur via les canaux sodiques, la désensibilisation des TRP, la modulation inflammatoire ou la signalisation cannabinoid périphérique sans exposition centrale substantielle. Une autre est celle des ligands CB1 biaisés ou à faible efficacité, qui visent à préserver la signalisation thérapeutique tout en réduisant l’intoxication. Une troisième est celle des composés à mécanisme mixte, souvent plus réalistes que le purisme d’une cible unique, où des actions modestes sur plusieurs nœuds de la douleur ou de l’anxiété battent un ligand « propre » mais cliniquement faible.
Le lien translationnel qui maintient ce domaine en vie est simple : la séparation de l’analgésie et du high semble possible, du moins dans les systèmes au stade de la recherche. Le résumé ScienceDaily de 2026 décrivant « a cannabis compound that relieves pain without the high » doit être traité avec prudence, car les titres dépassent les données, mais le concept correspond à l’orientation générale de la chimie médicinale. Il en va de même pour les travaux sur les canaux sodiques concernant THC. Il en va de même pour les efforts visant à construire des ligands sélectifs par docking et conception structurale. Le schéma est cohérent même lorsque certaines affirmations individuelles nécessitent des ajustements.
Ce à quoi le pipeline ne ressemble probablement pas, en revanche, est un succès clinique massif à court terme. La promiscuité des cibles, la complexité des métabolites, les différences entre espèces et les effets de formulation continuent de briser les histoires simples. Mais le domaine n’est plus bloqué à demander si les cannabinoid sont « vraiment » à propos de CB1 et CB2. Les développeurs de médicaments ont déjà répondu à cette question avec leurs budgets de chimie. Ils conçoivent pour l’espace au-delà de ces récepteurs, car c’est là que réside maintenant la meilleure chance de séparer le bénéfice de la toxicité.
Idées reçues courantes et controverses non résolues
La plus grande erreur dans les débats publics sur les cannabinoid est le réductionnisme récepteur : l’envie de forcer une pharmacologie complexe dans une cible vedette unique. Cette habitude a toujours été fragile, et elle devient plus risquée à mesure que les régulateurs et les développeurs de médicaments font face à des composés qui ne sont plus simplement du « THC » ou du « CBD » au sens simple, d’origine végétale. En 2025, HHS a déclaré que « 7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety » en soutenant une action de la DEA contre des produits 7-OH renforcés. Classe de médicament différente, même leçon : dès que des chimistes altèrent, enrichissent ou semi-synthétisent un squelette intoxicant, les anciennes hypothèses sur l’action des récepteurs et la sécurité peuvent échouer rapidement. La science du cannabis a le même problème. Un cannabinoid n’est pas une clé magique pour une seule serrure. C’est généralement un ligand promiscue dont les effets réels dépendent de la concentration, de l’exposition tissulaire, du métabolisme et des cibles hors récepteurs qui deviennent pertinentes à ces niveaux.
Existe-t-il vraiment un récepteur CB3
Il n’existe aucun récepteur CB3 faisant consensus.
La réponse paraît abrupte parce que les preuves l’exigent. Plusieurs récepteurs ont été proposés au fil des ans comme candidats « CB3 », en particulier GPR55, parfois GPR18, et occasionnellement d’autres GPCR orphelins qui répondent dans certains systèmes d’essai aux ligands cannabinoid ou aux lipides apparentés au système endocannabinoïde. Mais une cible proposée n’est pas une classe réceptrice acceptée. CB1 et CB2 ont mérité leur nom grâce à des preuves convergentes : clonage, pharmacologie reproductible des ligands, distribution tissulaire, signalisation et réplication large. Les candidats CB3 putatifs n’ont jamais franchi cette barre.
GPR55 est le suspect habituel. Il est exprimé dans le cerveau, les cellules immunitaires, l’intestin et l’os, et certains cannabinoid interagissent bel et bien avec lui. CBD a souvent été décrit comme antagoniste de GPR55 dans des essais cellulaires ; certains cannabinoid synthétiques montrent eux aussi une activité. Pourtant, la pharmacologie est incohérente d’un laboratoire à l’autre, d’un ligand à l’autre et d’une lecture à l’autre. Certains composés paraissent actifs dans un essai de signalisation et silencieux dans un autre. Les différences entre espèces compliquent le tableau. Les ligands endogènes sont débattus. Et surtout, appeler GPR55 « CB3 » suggère une place établie dans la famille canonique des récepteurs cannabinoid que le domaine ne lui a pas encore accordée.
C’est important parce que les étiquettes peuvent dépasser les preuves. Une fois qu’un récepteur reçoit un surnom accrocheur, ce surnom commence à faire un travail explicatif qu’il n’a pas gagné. Douleur ? CB3. Anxiété ? CB3. Effets osseux ? CB3. Ce n’est pas de la pharmacologie ; c’est du branding. La position plus prudente est que GPR55, GPR18, GPR119 et des récepteurs apparentés sont des cibles hors CB1/CB2 avec des degrés variables de preuves de modulation par les cannabinoid ou des lipides de type cannabinoid. Certains peuvent compter beaucoup dans des tissus spécifiques. Aucun n’a acquis le statut consensuel de « troisième récepteur cannabinoid ».
La découverte de médicaments a évolué en conséquence. Les articles de chimie médicinale n’agissent pas comme si un nouveau nom de récepteur allait résoudre le système. L’article de 2016 du Journal of Medicinal Chemistry « Library Docking for Cannabinoid-2 Receptor Ligands » est un bon marqueur de l’état réel du domaine : conception fondée sur la structure, sélectivité des récepteurs, ingénierie des squelettes et optimisation dirigée par la cible, pas mythologie autour d’un mystérieux CB3 censé tout expliquer. De même, les affirmations des entreprises concernant les composés cannabinoid de nouvelle génération mettent désormais souvent l’accent sur des mécanismes différenciés plutôt que sur un simple agonisme récepteur plus fort. Le communiqué préclinique de 2025 de MIRA Pharmaceuticals sur MIRA-55 affirmait explicitement un « differentiated mechanism of action » et une activité anxiolytique par rapport au THC. C’est du matériel promotionnel, pas de la science établie, mais cela reflète un véritable changement stratégique : les thérapeutiques cannabinoid utiles pourraient venir du fait de s’éloigner de l’intoxication CB1 brute, et non de la découverte d’un récepteur fourre-tout.
CBD agit-il principalement par la sérotonine
Là encore non, même si la signalisation sérotoninergique fait partie de l’histoire.
CBD est souvent présenté dans la culture populaire comme étant essentiellement un médicament naturel de 5-HT1A. Cette simplification persiste parce qu’elle repose sur de vraies données. Dans des études précliniques, CBD a montré des effets de type anxiolytique et anti-stress qui sont réduits par des antagonistes de 5-HT1A dans certains paradigmes. Des études expérimentales chez l’humain ont également suggéré que les mécanismes sérotoninergiques peuvent contribuer à des effets anxiolytiques aigus dans certaines conditions. Mais « contribue à » n’est pas « agit principalement par ».
CBD est pharmacologiquement large. Il a une faible affinité pour CB1 et CB2 comparée à THC, mais cela ne le transforme pas par défaut en agent sérotoninergique à cible unique. Dans la littérature, les contributeurs plausibles incluent TRPV1, 5-HT1A, GPR55, la signalisation de l’adénosine, PPAR-gamma, la gestion du calcium intracellulaire, le tonus endocannabinoïde lié à FAAH dans certains contextes et les effets de métabolites qui peuvent ne pas correspondre au composé parent. La concentration compte ici plus que beaucoup d’explications ne l’admettent. Un effet sur un récepteur observé in vitro à l’échelle micromolaire peut ne pas dominer chez l’humain après une dose orale standard, où l’absorption est variable et le métabolisme de premier passage important.
Le dossier clinique s’oppose aux affirmations mono-mécanistiques. L’usage FDA le plus solidement établi de CBD purifié ne concerne pas l’anxiété, mais les syndromes épileptiques : la solution orale est indiquée pour le syndrome de Lennox-Gastaut, le syndrome de Dravet et le complexe de sclérose tubéreuse chez les patients âgés d’un an et plus. Cet usage approuvé vous apprend déjà quelque chose. Si CBD était « principalement sérotoninergique », son profil thérapeutique le plus reproductible serait difficile à concilier avec ce que les cliniciens l’emploient réellement à traiter. La sérotonine peut compter dans les contextes anxieux, surtout pour les réponses liées à 5-HT1A, mais la pharmacologie humaine de CBD ne se résume pas à une étiquette sérotoninergique.
Même dans l’anxiété, le mécanisme varie probablement avec la dose et le contexte. TRPV1 en est un bon exemple. CBD peut activer TRPV1, et la signalisation TRP est suffisamment centrale à la biologie sensorielle pour que David Julius et Ardem Patapoutian aient reçu le prix Nobel 2021 pour leurs découvertes sur les récepteurs de la température et du toucher. Pourtant, les effets TRPV1 ne sont pas linéaires. L’activation peut être suivie d’une désensibilisation ; des doses faibles et fortes peuvent produire des issues comportementales différentes. Donc, lorsque quelqu’un dit « CBD agit par la sérotonine », la bonne réponse est : en partie, parfois, et probablement pas seul.
Un seul cible peut-il expliquer un effet de plante entière
Aucune cible unique ne peut expliquer un effet de cannabis entier, et essayer de forcer une telle explication obscurcit généralement plus qu’il n’éclaire.
Les effets de plante entière émergent de variables empilées. Commencez par la composition : THC, CBD, minor cannabinoid tels que CBG, CBC, THCV, précurseurs acides, produits d’oxydation et métabolites apportent chacun des profils de cibles distincts. Ajoutez la voie d’administration, et le tableau change à nouveau. Les cannabinoid inhalés atteignent rapidement le cerveau ; les produits oraux subissent un métabolisme de premier passage et génèrent des espèces actives différentes. La loi américaine définit encore le hemp par un seuil de delta-9 THC de « not more than 0.3 percent on a dry weight basis », mais cette limite légale dit peu de choses sur la pharmacologie de tout le reste de l’échantillon ou sur ce qui se forme après le métabolisme.
Ajoutez ensuite la spécificité tissulaire. Un cannabinoid peut affecter en parallèle CB1 central, les canaux TRP périphériques, la signalisation immunitaire et les récepteurs nucléaires. Le rapport 2025 de l’Université hébraïque selon lequel THC inhibe les nocicepteurs périphériques en ciblant NaV1.7 et NaV1.8 est un exemple saisissant, parce qu’il brise l’équation paresseuse « effet THC=effet CB1 ». Si même THC possède des actions significatives sur les canaux sodiques dans les voies de la douleur, l’idée qu’une fleur, un extrait ou un comestible ait un seul récepteur maître devient difficile à défendre. La recherche 2026 mise en avant par ScienceDaily — un cannabinoid soulageant la douleur sans le high — va dans le même sens, même si elle reste au stade de la recherche. L’analgésie peut être dissociée de l’intoxication centrale en exploitant la restriction périphérique ou des cibles hors CB1. C’est là que le domaine se dirige.
L’attente et la biologie individuelle comptent aussi. L’expérience antérieure, le niveau d’anxiété, la génétique, le sexe, l’activité enzymatique hépatique, le sommeil, l’inflammation et les médicaments concomitants modifient tous ce que ressent un produit donné et ce qu’il fait physiologiquement. Les terpènes peuvent contribuer dans certains cas, mais ils ne doivent pas être traités comme des chefs d’orchestre magiques de l’effet total. Leurs concentrations sont souvent faibles, et les preuves humaines sont plus minces que ne le suggère le langage marketing.
La vision plus dure mais plus exacte est la suivante : les effets du cannabis sont émergents. Ils naissent de nombreuses interactions modestes, pas d’un récepteur-slogan. Cela rend la science moins propre. Cela la rend aussi plus honnête.
Interprétation pratique pour les lecteurs, les cliniciens et les chercheurs
La leçon pratique de la pharmacologie cannabinoid hors CB1/CB2 est simple mais exigeante : les affirmations mécanistiques devraient recevoir un examen plus rigoureux, et non une acceptation plus facile, dès qu’un article invoque les canaux TRP, les PPAR, GPR55, 5-HT1A, la signalisation adénosinergique ou les canaux NaV. Les cannabinoid sont souvent des molécules pharmacologiquement promiscues. Cela peut être utile en découverte de médicaments. Cela peut aussi induire les lecteurs en erreur en leur faisant prendre n’importe quel résultat de cible dans une boîte pour l’explication d’un effet clinique.
Une bonne règle consiste à classer les preuves selon leur distance au patient. Un essai de liaison est un point de départ, pas une conclusion. Les études de signalisation cellulaire viennent ensuite. Les travaux chez l’animal peuvent affiner la plausibilité. La pharmacologie expérimentale chez l’humain compte davantage. Les preuves issues d’un médicament approuvé comptent le plus, et même là, l’étiquette ne règle pas toujours le mécanisme. La solution orale de cannabidiol, par exemple, est approuvée par la FDA pour les crises associées au syndrome de Lennox-Gastaut, au syndrome de Dravet et au complexe de sclérose tubéreuse chez les patients âgés d’un an et plus, alors que son profil thérapeutique n’est pas expliqué proprement par l’activation de CB1 ou CB2. C’est précisément pourquoi les affirmations sur TRPV1, GPR55, 5-HT1A et les cibles intracellulaires reviennent sans cesse.
Comment lire les affirmations mécanistiques sur les cannabinoid de façon critique
Commencez par l’espèce et le système. L’effet a-t-il été démontré dans du tissu humain, une lignée cellulaire de rongeur, des ovocytes de Xenopus ou des récepteurs surexprimés dans des HEK293 ? Ce n’est pas interchangeable. GPR55 en est un bon exemple : une étude peut montrer que CBD agit comme antagoniste dans un système recombinant, tandis qu’un autre contexte produit une signalisation faible ou variable parce que l’expression du récepteur, les lipides endogènes et la conception de l’essai diffèrent. Appeler cela « le mécanisme » est généralement prématuré.
Ensuite, posez la question de la concentration. C’est là que beaucoup d’affirmations sur les cannabinoid s’effondrent. Un article peut rapporter une activation de TRPV1, une transactivation de PPAR-gamma ou une inhibition des canaux sodiques à des concentrations micromolaires. Très bien. Mais la dose humaine dont il est question produit-elle vraiment des concentrations tissulaires libres de cet ordre ? L’administration orale de cannabinoid fait face au métabolisme de premier passage, à une liaison protéique importante et à une distribution tissulaire inégale. Une interaction à 30 micromolaires in vitro peut être une chimie intéressante et une pharmacologie clinique sans intérêt. L’inverse peut aussi arriver : une accumulation locale dans les tissus, des métabolites actifs ou le partitionnement lipidique peuvent rendre plus plausible une cible intracellulaire que ne le laissent penser les niveaux plasmatiques seuls. Dans les deux cas, la concentration n’est pas un détail. C’est la question.
La mesure directe de la cible compte aussi. Les chercheurs ont-ils réellement bloqué l’effet avec un antagoniste sélectif, réduit l’expression du récepteur ou mesuré le courant du canal ? Ou bien ont-ils inféré le mécanisme à partir de ressemblances avec la littérature antérieure ? Pour les canaux TRP, surtout TRPV1 et TRPA1, cela importe parce que l’activation peut être biphasique et conduire à une désensibilisation. Un composé peut d’abord activer un canal puis réduire la réactivité ultérieure, ce qui signifie que « agoniste » ne se traduit pas toujours proprement par « plus de douleur » ou « plus de sensation de chaleur ». C’est l’une des raisons pour lesquelles le prix Nobel 2021 attribué à David Julius et Ardem Patapoutian, « for their discoveries of receptors for temperature and touch », est si pertinent ici : la signalisation somatosensorielle est mécaniquement riche, et les étiquettes réceptrices simplistes échouent souvent.
L’histoire NaV montre à quoi ressemble une preuve plus solide. Les chercheurs de l’Université hébraïque ont rapporté en 2025 que THC inhibe les nocicepteurs périphériques en ciblant les canaux sodiques nociceptifs NaV1.7 et NaV1.8. Cette affirmation est plus informative que des déclarations vagues selon lesquelles THC « agit en dehors de CB1 », car elle nomme des canaux pertinents pour la douleur déjà centraux dans la recherche sur l’analgésie. Elle reformule aussi une hypothèse ancienne. Même un composé célèbre pour son intoxication médiée par CB1 peut avoir des actions non CB significatives, surtout dans les tissus périphériques.
Les lecteurs doivent aussi surveiller les explications concurrentes. Supposons qu’une étude relie CBD à l’anxiolyse via 5-HT1A. Hypothèse raisonnable. Mais la sédation a-t-elle été exclue ? Une modulation CB1 indirecte a-t-elle été exclue ? L’expérience a-t-elle distingué les effets au niveau du récepteur des changements de tonus endocannabinoïde, de la recapture de l’adénosine, de la signalisation inflammatoire ou de l’attente chez les sujets humains ? Les médicaments à cibles multiples annoncent rarement quelle cible fait l’essentiel du travail dans un modèle donné.
Le climat réglementaire actuel rend ce scepticisme plus qu’académique. En 2025, HHS a déclaré que « 7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety » en soutenant une action de classement sur des produits 7-OH renforcés. Ce n’est pas un cas de cannabis, mais la leçon est limpide : les régulateurs distinguent de plus en plus les constituants végétaux familiers des intoxicants renforcés, semi-synthétiques ou autrement modifiés ayant des profils de puissance et de sécurité différents. Une politique cannabinoid qui traite tous les composés comme des versions mises à l’échelle du delta-9-THC est pharmacologiquement obsolète.
Quelles questions les cliniciens devraient poser sur la pertinence des cibles
Les cliniciens n’ont pas besoin de mémoriser tous les GPCR orphelins pour bien interpréter les affirmations sur les cannabinoid. Ils ont besoin d’une check-list disciplinée.
Premièrement : l’effet a-t-il été démontré chez l’humain ou seulement dans des cellules et chez l’animal ? Un modèle inflammatoire murin peut soutenir la plausibilité d’une implication de PPAR-gamma ou de TRPA1, mais il n’établit pas un bénéfice patient. Deuxièmement : à quelles concentrations ou doses ? Si une cible proposée n’est engagée qu’à des niveaux supérieurs à ceux atteints par un dosage oral standard, ce mécanisme peut ne pas expliquer les résultats cliniques routiniers. Troisièmement : la cible a-t-elle été mesurée directement ? L’occupation réceptrice, l’électrophysiologie, le retour par antagoniste ou la perturbation génétique comptent tous davantage que l’inférence narrative.
Quatrièmement : la voie d’administration rend-elle l’affirmation plus ou moins crédible ? L’inhalation, l’ingestion orale, l’administration transdermique et l’application topique produisent des profils d’exposition très différents. Un mécanisme analgésique périphérique est plus plausible pour un composé topique ou à restriction périphérique que pour une molécule qui inonde rapidement le cerveau. Cette distinction compte si l’objectif thérapeutique est l’analgésie sans intoxication.
Cinquièmement : existe-t-il des explications concurrentes liées à des effets indésirables connus ? Si un patient rapporte moins d’anxiété après une préparation à base de cannabinoid, s’agit-il d’un effet anxiolytique médié par 5-HT1A, d’une réduction de la douleur, d’une sédation non spécifique ou d’une attente ? Si les marqueurs inflammatoires bougent, PPAR-gamma en est-il le moteur probable, ou des changements métaboliques ou immunitaires plus larges se sont-ils produits en amont ? Le mécanisme ne doit pas être déduit d’une simple amélioration symptomatique.
Pour les cliniciens, les minor cannabinoid appellent la même prudence. CBC, CBG, THCV et les cannabinoid acides sont souvent présentés comme s’ils possédaient chacun une signature cible stable. La littérature ne justifie pas encore cette confiance. Certains sont prometteurs. Aucun ne doit être considéré comme pharmacologiquement établi simplement parce qu’une marque ou un fil sur les réseaux sociaux lui attribue une spécificité réceptrice.
Où le domaine se dirige probablement ensuite
L’orientation la plus crédible à court terme est l’analgésie périphérique. L’attrait translationnel est évident : séparer le soulagement de la douleur de l’intoxication centrale. Le résumé ScienceDaily de 2026 décrivant des travaux sur « a cannabis compound that relieves pain without the high » doit être lu comme un signal au stade de la recherche, pas comme une réponse clinique achevée, mais il capture l’objectif de la chimie médicinale. Il en va de même pour les travaux NaV1.7/NaV1.8 de l’Université hébraïque : la biologie de la douleur pousse la science cannabinoid vers les nerfs périphériques, les canaux ioniques et une exposition sélective des tissus.
Les ligands des récepteurs nucléaires anti-inflammatoires constituent une autre voie forte. Les affirmations concernant PPAR-gamma nécessitent de meilleures données d’engagement de cible chez l’humain, mais le concept est suffisamment plausible pour justifier un développement sérieux, en particulier là où les voies métaboliques et inflammatoires se rejoignent. Les anxiolytiques à mécanisme multiple resteront aussi un territoire actif. MIRA Pharmaceuticals a déclaré dans un communiqué Nasdaq de 2025 que son candidat MIRA-55 présentait un « differentiated mechanism of action » et une « anxiolytic activity relative to THC » dans des données précliniques. Parce qu’il s’agit d’un rapport d’entreprise et préclinique, il doit être traité avec prudence. Néanmoins, il reflète une tendance réelle : les chercheurs ne se satisfont plus de demander si un candidat est « comme THC » ou « comme CBD ». Ils veulent des profils cibles définis.
Ce même changement est visible en chimie médicinale. L’article de 2016 du Journal of Medicinal Chemistry « Library Docking for Cannabinoid-2 Receptor Ligands » signalait une approche fondée sur la structure qui n’a cessé de se développer depuis : construire des ligands pour des cibles spécifiques, des tissus spécifiques et des biais de signalisation spécifiques. Les minor cannabinoid mieux définis émergeront probablement de cette vision, et non d’affirmations générales selon lesquelles chaque cannabinoid rare occupe une niche bien-être unique.
La leçon pratique est nette. En lisant la pharmacologie cannabinoid, posez cinq questions à chaque fois : L’effet a-t-il été montré chez l’humain ou seulement dans des cellules ? À quelles concentrations ? La cible a-t-elle été mesurée directement ? La dose atteint-elle cette cible dans un tissu vivant ? Existe-t-il des explications concurrentes ? L’avenir de la science cannabinoid n’est pas une pharmacologie moins spécifique, mais une pharmacologie plus spécifique.
Références
- [1]HHS and FDA support DEA action on dangerous 7-OH products. HHS Press Room, 2025. https://www.hhs.gov/press-room/hhs-fda-support-dea-7-oh-scheduling.html
- [2]EPIDIOLEX (cannabidiol) oral solution label. FDA drug label, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/210365s016lbl.pdf
- [3]Library Docking for Cannabinoid-2 Receptor Ligands. Journal of Medicinal Chemistry, 2016. https://pubs.acs.org/doi/10.1021/acs.jmedchem.6c00835
- [4]MIRA Pharmaceuticals Reports New Preclinical Data Demonstrating MIRA-55's Differentiated Mechanism of Action and Anxiolytic Activity Relative to THC. Nasdaq press release, 2025. https://www.nasdaq.com/press-release/mira-pharmaceuticals-reports-new-preclinical-data-demonstrating-mira-55s
- [5]Psychoactive cannabinoid THC inhibits peripheral nociceptors by targeting NaV1.7 and NaV1.8 nociceptive sodium channels. Research portal summary, 2025. https://cannabinoids.huji.ac.il/publications/psychoactive-cannabinoid-thc-inhibits-peripheral-nociceptors-targeting
- [6]A cannabis compound that relieves pain without the high. ScienceDaily, 2026. https://www.sciencedaily.com/releases/2026/06/260619033343.htm
- [7]The Nobel Prize in Physiology or Medicine 2021. Nobel Prize Press Release, 2021. https://www.nobelprize.org/prizes/medicine/2021/press-release/
- [8]Agriculture Improvement Act of 2018. Congress.gov, 2018. https://www.congress.gov/bill/115th-congress/house-bill/2/text