






Sommario
- Perché la scienza dei cannabinoid non può essere ridotta a CB1 e CB2
- Il sistema endocannabinoid rispetto al più ampio panorama dei target dei cannabinoid
- Canali TRP: i sensori di calore, dolore e irritazione su cui i cannabinoid continuano a intervenire
- PPAR: i cannabinoid come segnali lipidici intracellulari, non solo come ligandi dei recettori di membrana
- GPR55, GPR18, GPR119 e il problema dei GPCR orfani
- Segnalazione della serotonina: dove i cannabinoid si intersecano con i sistemi 5-HT
- Oltre l’elenco richiesto: canali del sodio e altri target non canonici che stanno già cambiando il dibattito sul dolore
- In cosa differiscono i cannabinoid specifici quando si smette di chiedere solo di CB1 e CB2
- I metodi contano: perché la progettazione dei saggi modella ciò che pensiamo facciano i cannabinoid
- Livelli di evidenza: dal piatto cellulare alla clinica
- Sicurezza, regolamentazione e perché la farmacologia off-target conta per la salute pubblica
- Drug discovery: progettare cannabinoid e molecole ispirate ai cannabinoid per target non CB1/CB2
- Convinzioni errate comuni e controversie irrisolte
- Interpretazione pratica per lettori, clinici e ricercatori
Perché la scienza dei cannabinoid non può essere ridotta a CB1 e CB2
La versione abbreviata della farmacologia dei cannabinoid dice questo: THC agisce su CB1, gli effetti immunitari passano attraverso CB2, e tutto il resto è una nota a piè di pagina. Questa impostazione è facile da insegnare e facile da ripetere. È anche sbagliata abbastanza spesso da ostacolare una reale comprensione di dolore, infiammazione, ansia, prurito, nausea, metabolismo e neuroprotezione.
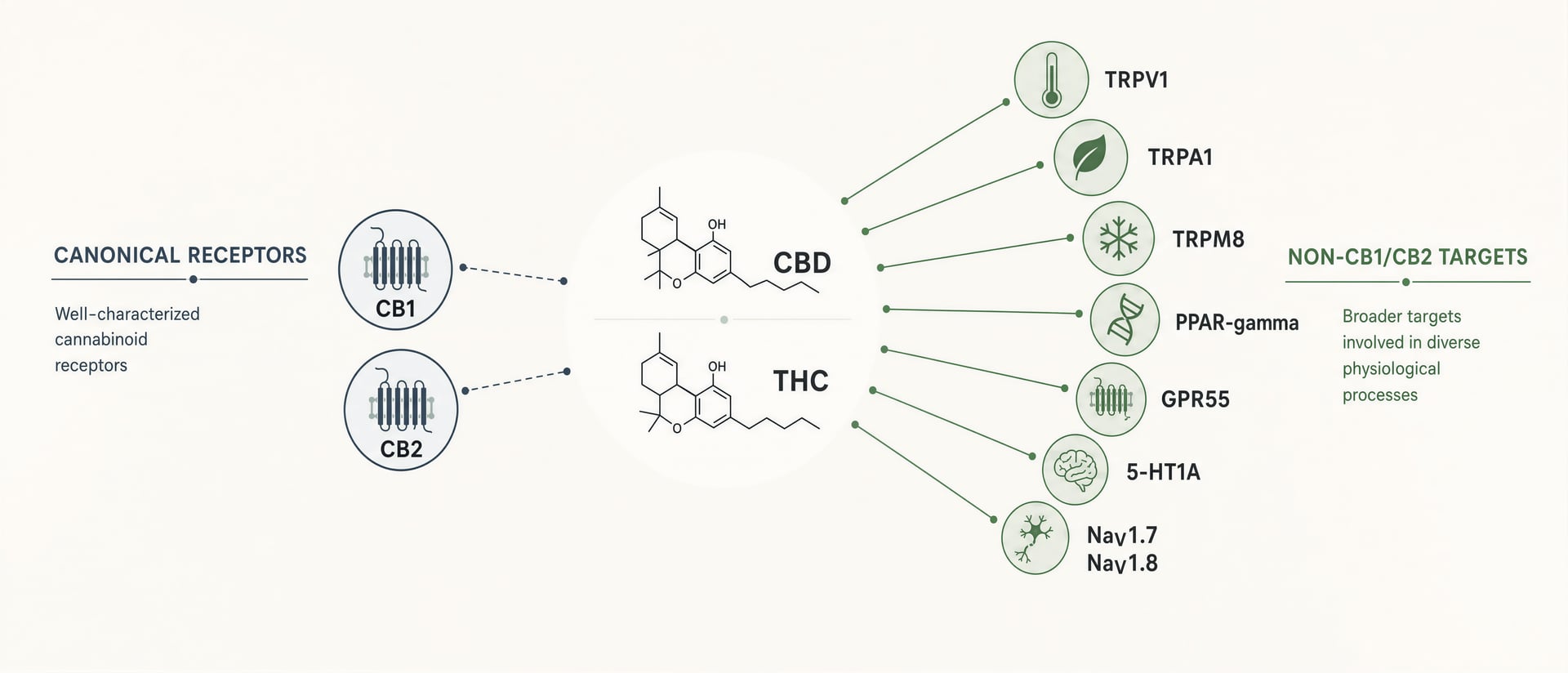
CB1 e CB2 contano. CB1 è abbondante nel cervello e spiega gran parte dell’intossicazione da THC, dell’alterazione della memoria, degli effetti sull’appetito e di una parte della sua analgesia. CB2 è centrale in molte discussioni su immunità e infiammazione. Ma i cannabinoid non sono ligandi ordinati costruiti per un solo recettore ciascuno. Sono molecole lipofile, flessibili nella forma, che interagiscono con un campo farmacologico più ampio: canali recettoriali transitori come TRPV1 e TRPA1, recettori nucleari come PPAR-gamma, GPCR orfani o ancora discussi come GPR55 e GPR18, recettori della serotonina incluso 5-HT1A, segnalazione correlata all’adenosina, trasporto e metabolismo degli acidi grassi e, in lavori più recenti, canali del sodio voltaggio-dipendenti tra cui NaV1.7 e NaV1.8.[1]HHS and FDA support DEA action on dangerous 7-OH products. U.S. Department of Health and Human Services. HHS Press Room, 2025. https://www.hhs.gov/press-room/hhs-fda-support-dea-7-oh-scheduling.html
Questo campo più ampio conta perché il meccanismo determina rischio e beneficio. I regolatori stanno già affrontando questo problema in battaglie normative affini. Nel 2025, il Department of Health and Human Services degli Stati Uniti ha affermato che “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” sostenendo l’azione della DEA sui prodotti 7-OH potenziati. Il composto specifico non è un cannabinoid, ma la lezione politica è trasferibile: quando i chimici iniziano a modificare gli scheletri dei prodotti naturali e a concentrare i metaboliti, le semplici categorie basate sulla fonte smettono di proteggere il pubblico. Il profilo dei target di una molecola conta più del fatto che la divulgazione popolare la consideri familiare.
Il mito del recettore nella divulgazione popolare sul cannabis
Le spiegazioni popolari sul cannabis presentano di solito i recettori come interruttori on-off: THC accende CB1, CBD non “si lega fortemente”, quindi CBD deve essere debole o misterioso. Questo racconto fonde in un solo verbo vago, bind, più idee farmacologiche distinte.
L’agonismo ortosterico è il caso classico. Un ligando occupa il sito attivo principale del recettore e stabilizza la segnalazione. THC è un agonista parziale di CB1 e CB2. Questo è un tipo di azione, non il modello di tutta la biologia dei cannabinoid. Un composto può invece agire allostericamente, modificando il modo in cui un altro ligando funziona sul recettore senza occupare lo stesso sito. Può aprire, sensibilizzare o desensibilizzare un canale ionico. Può entrare nella cellula e attivare un recettore nucleare che cambia la trascrizione genica nell’arco di ore anziché di millisecondi. Può inibire un trasportatore, alterare le proprietà della membrana o rallentare un enzima che degrada un lipide segnale endogeno.[2]EPIDIOLEX (cannabidiol) oral solution label. U.S. Food and Drug Administration. FDA drug label, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/210365s016lbl.pdf
CBD è la confutazione più chiara del riduzionismo recettoriale. Il suo uso clinico approvato non si basa sull’agonismo di CB1. L’etichetta FDA della soluzione orale di cannabidiol indica che è prescritta per convulsioni associate alla sindrome di Lennox-Gastaut, alla sindrome di Dravet e al complesso della sclerosi tuberosa in pazienti di età pari o superiore a 1 anno. Qualunque sia il meccanismo completo alla base di tale effetto, esso non è spiegato in modo adeguato dal vecchio racconto secondo cui un’azione cannabinoid significativa equivale a una forte attivazione di CB1 o CB2. Tra i candidati meccanicistici richiamati ripetutamente in letteratura vi sono TRPV1, segnalazione correlata a 5-HT1A, modulazione dell’adenosina, effetti sul calcio intracellulare e interazioni con enzimi o trasportatori. Nessuno può essere considerato la risposta unica, ma insieme mostrano perché il mito semplicistico del recettore fallisce.
Anche la storia va nella stessa direzione. Il lavoro sugli endocannabinoid di Raphael Mechoulam ha aperto un campo centrato su anandamide e 2-AG, eppure anche anandamide non è soltanto un ligando di CB1. Attiva anche TRPV1, il recettore del calore e della capsaicina la cui più ampia importanza sensoriale è stata riconosciuta nel premio Nobel per la Fisiologia o Medicina 2021 a David Julius e Ardem Patapoutian per le scoperte dei recettori per temperatura e tatto. Una volta che un endocannabinoid endogeno può segnalare attraverso sia un GPCR sia un canale TRP, il modello “solo CB1/CB2” non è più un modello. È una caricatura.
Policomplesso farmacologico: un ligando, molti target
Un punto di partenza migliore è la policomplessità farmacologica. Un ligando, molti target, con diverse affinità, efficacie, tessuti e conseguenze. In farmacologia, “sporco” è talvolta usato in senso peggiorativo, ma per i cannabinoid è spesso semplicemente descrittivo.
Si consideri quanti tipi di azione rientrano sotto lo stesso termine ombrello. THC è un agonista parziale di CB1/CB2, eppure lavori del 2025 evidenziati dalla Hebrew University hanno riportato che THC inibisce i nocicettori periferici prendendo di mira i canali del sodio nocicettivi NaV1.7 e NaV1.8. Non si tratta affatto di agonismo recettoriale. È inibizione di canali ionici su target già considerati candidati primari per farmaci antalgici. Se questa linea di lavoro reggerà in più specie e condizioni di dosaggio, parte dell’analgesia da THC potrebbe derivare da un meccanismo più simile a un freno locale dell’eccitabilità che a un classico effetto sui recettori cannabinoid.
CBD mostra uno stile diverso di promiscuo. In diversi sistemi di saggio è stato segnalato come influente su TRPV1, TRPA1, TRPM8, 5-HT1A, PPAR-gamma, GPR55 e tono dell’adenosina, tra gli altri. Il problema non è la mancanza di meccanismi. Il problema è capire quali meccanismi contino a concentrazioni clinicamente raggiungibili nell’uomo. L’impegno del target in vitro è economico. La traslazione è difficile. Un effetto micromolare in una linea cellulare che sovraesprime un recettore non spiega automaticamente gli esiti nei pazienti dopo somministrazione orale, metabolismo di primo passaggio, legame alle proteine e partizione tissutale.
Altri fitocannabinoid complicano ulteriormente il quadro. In alcuni sistemi CBG è stato discusso come composto attivo su alfa-2 adrenergici, TRP e 5-HT1A. CBC è stato collegato ai canali TRPA1 e TRPV. THCV può comportarsi in modo diverso dal delta-9-THC su CB1 a seconda della dose e del contesto, pur presentando anche possibilità non CB1. I cannabinoid acidi come CBDA e THCA sollevano ulteriori interrogativi perché decarbossilazione, stabilità e formazione di metaboliti alterano tutti l’esposizione ai target. La stessa etichetta di una bottiglia può quindi nascondere una farmacologia molto diversa quando entrano in gioco via di somministrazione, calore, metabolismo e formulazione.[3]Library Docking for Cannabinoid-2 Receptor Ligands. American Chemical Society. Journal of Medicinal Chemistry, 2016. https://pubs.acs.org/doi/10.1021/acs.jmedchem.6c00835
Anche all’interno della farmacologia GPCR, il campo è andato oltre le etichette grossolane. GPR55 è talvolta ancora chiamato candidato “CB3”, ma ciò resta contestato per buone ragioni; segnalazione, set di ligandi e ruolo fisiologico non si sovrappongono in modo pulito ai recettori cannabinoid classici. Anche GPR18 e GPR119 sono discussi nella letteratura adiacente ai cannabinoid, soprattutto per infiammazione, metabolismo e segnalazione intestinale, ma le evidenze sono disomogenee. I chimici medicinali lo sanno. Un articolo del 2016 sul Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, ha catturato un approccio basato sulla struttura che è quasi l’opposto della lore popolare sui recettori: progettazione selettiva del target, docking, ottimizzazione dello scheletro e separazione intenzionale degli effetti desiderati da quelli indesiderati. Il campo non sta chiedendo “colpisce i recettori cannabinoid?” Sta chiedendo quali target, in quale stato, in quale tessuto, a quale concentrazione e con quale bias.
Perché i target non CB1/CB2 contano clinicamente
È qui che la scienza smette di essere semantica e inizia a incidere sulla medicina.
Per il dolore, i target non CB1 potrebbero essere la via più plausibile per farmaci utili con meno intossicazione. TRPV1, TRPA1, canali del sodio periferici e vie trascrizionali infiammatorie offrono tutti modi per ridurre l’attivazione dei nocicettori o la sensibilizzazione neuroimmune senza una forte attivazione centrale di CB1. Un report del 2026 di ScienceDaily su un composto del cannabis che “relieves pain without the high” è soltanto un segnale allo stadio di ricerca, non una risposta clinica definitiva, ma la direzione ha senso. Se l’analgesia può essere spostata verso canali ionici periferici o un’esposizione tissutale ristretta, il vecchio compromesso tra sollievo dal dolore e carico psicoattivo può attenuarsi.
Per infiammazione e metabolismo, PPAR-gamma è un buon esempio di perché le categorie recettoriali contino. I PPAR sono recettori nucleari, non recettori cannabinoid di membrana. La loro attivazione modifica programmi di espressione genica coinvolti nella gestione dei lipidi, nella sensibilità all’insulina e nel tono infiammatorio. Alcuni effetti dei cannabinoid riportati in modelli metabolici o infiammatori si inseriscono meglio in questa biologia trascrizionale lenta che nella segnalazione rapida di CB1. Ma ancora una volta, concentrazione e accesso intracellulare contano. Un articolo che mostri l’attivazione di PPAR in un saggio reporter non dimostra un effetto anti-infiammatorio clinicamente rilevante nell’uomo.[4]MIRA Pharmaceuticals Reports New Preclinical Data Demonstrating MIRA-55's Differentiated Mechanism of Action and Anxiolytic Activity Relative to THC. MIRA Pharmaceuticals. Nasdaq press release, 2025. https://www.nasdaq.com/press-release/mira-pharmaceuticals-reports-new-preclinical-data-demonstrating-mira-55s
Per ansia e nausea, i meccanismi legati alla serotonina ricompaiono di continuo, soprattutto 5-HT1A. I dati sono misti e spesso indiretti, ma la persistenza del segnale è significativa. La reputazione ansiolitica di CBD è difficile da mappare solo su CB1/CB2. Questa è una delle ragioni per cui le aziende stanno cercando di progettare composti ispirati ai cannabinoid ma differenziati, invece di realizzare semplicemente analoghi più forti del THC. Nel 2025, MIRA Pharmaceuticals ha riportato che il proprio candidato MIRA-55 mostrava un “differentiated mechanism of action” e “anxiolytic activity relative to THC”. I comunicati aziendali sono prove di basso livello e vanno trattati come tali. Tuttavia, indicano la direzione del drug development: lontano dall’idea che il miglior farmaco cannabinoid sia soltanto una stimolazione CB1 più pulita.
Prurito, emicrania, epilessia, disturbi intestinali e neuroprotezione si collocano tutti nella stessa zona meccanicistica. I canali TRP regolano il guadagno sensoriale. I GPCR orfani possono modellare la segnalazione immunitaria ed epiteliale. I PPAR alterano i programmi infiammatori. I canali del sodio controllano direttamente l’eccitabilità. Le vie della serotonina influenzano ansia, emesi e risposte allo stress. Una volta che questi sistemi vengono posti accanto a CB1 e CB2, anziché sotto di essi, molti effetti reali dei cannabinoid appaiono meno misteriosi e più farmacologicamente ordinari.
Il modello semplificato sopravvive perché è facile. Il modello migliore sopravvive al confronto con i dati.
Il sistema endocannabinoid rispetto al più ampio panorama dei target dei cannabinoid
La divulgazione sul cannabis tratta spesso la farmacologia come una storia a due recettori: CB1 spiega gli effetti psicoattivi, CB2 spiega gli effetti immunitari e tutto il resto è dettaglio. Questo quadro è troppo piccolo rispetto alle evidenze. Non spiega perché cannabidiol non possa essere chiaramente ricondotto a CB1 o CB2, perché alcuni cannabinoid scatenino bruciore o analgesia attraverso i canali TRP, perché i recettori nucleari intracellulari come PPAR-γ continuino ad apparire negli studi sull’infiammazione e perché persino THC possa influire su canali del sodio rilevanti per il dolore al di fuori della segnalazione cannabinoid classica. Se il campo vuole spiegare dolore, ansia, infiammazione, controllo delle crisi epilettiche o problemi di sicurezza con nuovi composti intossicanti, il riduzionismo recettoriale deve essere abbandonato.
Il momento regolatorio lo rende evidente. Nel 2025, HHS ha affermato che “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” sostenendo un intervento di scheduling contro prodotti 7-OH potenziati. La dichiarazione non riguardava il cannabis, ma coglie la stessa lezione farmacologica: quando i produttori passano da costituenti vegetali familiari a intossicanti potenziati, semi-sintetici o strutturalmente modificati, le semplici etichette di categoria smettono di essere utili. “THC-like” dice molto meno del profilo dei target, della potenza, dei metaboliti, della distribuzione tissutale e dell’attività off-target.
Target canonici: CB1, CB2, anandamide e 2-AG
Il sistema endocannabinoid canonico conta ancora. CB1 e CB2 sono recettori accoppiati a proteine G, principalmente Gi/o, identificati alla fine del XX secolo e mappati in dettaglio da ricercatori come Ken Mackie e Vincenzo Di Marzo. CB1 è fortemente espresso nel sistema nervoso centrale, soprattutto in corteccia, ippocampo, gangli della base e cervelletto, motivo per cui l’agonismo parziale di THC lì è collegato a intossicazione, effetti sulla memoria, alterazioni del controllo motorio e cambiamenti dell’appetito. CB2 è arricchito nelle cellule immunitarie e nei tessuti periferici, pur non essendo assente nel cervello. L’attivazione di uno dei due recettori di solito riduce la formazione di cAMP, modula i canali ionici e cambia il rilascio di trasmettitori.
I ligandi endogeni sono anandamide e 2-arachidonoylglycerol, di solito abbreviati in anandamide e 2-AG. Il gruppo di Raphael Mechoulam è stato centrale in questa storia: anandamide fu identificata nel 1992, 2-AG poco dopo. Non sono immagazzinati nelle vescicole sinaptiche come i neurotrasmettitori classici. Sono sintetizzati “su richiesta” da precursori lipidici di membrana e spesso agiscono in modo retrogrado, spostandosi dalle cellule postsinaptiche ai terminali presinaptici per smorzare il rilascio di neurotrasmettitori. Anandamide viene degradata soprattutto da FAAH; 2-AG soprattutto da MAGL. Questo ciclo biochimico è la spina dorsale del sistema endocannabinoid.
Ma la spina dorsale non è tutto lo scheletro. Anandamide è anche un agonista di TRPV1. CBD ha una bassa affinità diretta per CB1 e CB2 rispetto a THC ma ha chiaramente azioni clinicamente significative; la soluzione orale di cannabidiol approvata dalla FDA è indicata per convulsioni associate alla sindrome di Lennox-Gastaut, alla sindrome di Dravet e al complesso della sclerosi tuberosa in pazienti di 1 anno e oltre. Questo uso approvato ricorda costantemente che gli effetti clinicamente rilevanti dei cannabinoid non devono per forza coincidere con una forte agonismo di CB1.
Cosa conta come target dei cannabinoid
Una definizione pratica è migliore di una purista. Un target dei cannabinoid è qualsiasi sito molecolare al quale un fitocannabinoid, un endocannabinoid, un metabolita o uno scheletro ispirato ai cannabinoid si lega o modula funzionalmente la segnalazione a concentrazioni che potrebbero essere rilevanti in cellule, tessuti, animali o esseri umani. Secondo questo standard, il panorama si amplia rapidamente.
I canali TRP sono gli esempi non CB più familiari. TRPV1, TRPA1, TRPV2 e TRPM8 ricorrono nei lavori sui cannabinoid. Non è una nota marginale. David Julius e Ardem Patapoutian hanno condiviso il premio Nobel per la Fisiologia o Medicina 2021 “for their discoveries of receptors for temperature and touch”, un richiamo al fatto che i canali ionici che governano calore, freddo, irritazione e meccanosensazione siedono direttamente nelle vie del dolore. Anandamide attiva TRPV1. CBD, CBG, CBC e i cannabinoid acidi hanno tutti mostrato attività su TRP in vitro, spesso con effetti dipendenti dalla concentrazione e talvolta bifasici. Un cannabinoid che inizialmente attiva TRPV1 può poi desensibilizzarlo, producendo il paradosso di un’irritazione iniziale seguita da analgesia.
I PPAR allargano ulteriormente il quadro. PPAR-α e PPAR-γ sono recettori nucleari che regolano la trascrizione relativa al metabolismo lipidico e all’infiammazione. Alcuni cannabinoid e lipidi correlati agli endocannabinoid agiscono qui direttamente o dopo accumulo e metabolismo intracellulari. Si tratta di effetti trascrizionali lenti, non della segnalazione in millisecondi di CB1. Questo è importante per le affermazioni sull’infiammazione cronica, che spesso hanno più senso attraverso la segnalazione nucleare che attraverso l’attività acuta del recettore cannabinoid a livello sinaptico.
Poi ci sono i GPCR orfani o ancora discussi, in particolare GPR55, GPR18 e GPR119. GPR55 è stato più volte proposto come candidato “CB3”, e l’etichetta resta prematura. Il recettore esiste; la classificazione è contestata. CBD è spesso descritto come antagonista o modulatore negativo di GPR55 in sistemi sperimentali, mentre alcuni lipidi endogeni e ligandi sintetici possono attivarlo. GPR18 e GPR119 ricorrono in infiammazione, metabolismo e segnalazione immunitaria, ma le evidenze sono disomogenee e gli effetti di specie possono essere notevoli.
Anche i recettori della serotonina, soprattutto 5-HT1A, appartengono a questa mappa più ampia. La letteratura ansiolitica e antiemetica di CBD spesso implica 5-HT1A, anche se si discute ancora se si tratti di agonismo diretto o di facilitazione indiretta. Questa distinzione conta. Un composto che si lega debolmente a un recettore ma sposta in modo affidabile il comportamento del circuito attraverso meccanismi allosterici o di rete può comunque avere effetti in vivo rilevanti. La stessa cautela si applica ai programmi preclinici riportati dalle aziende: nel 2025, MIRA Pharmaceuticals ha affermato che il proprio candidato MIRA-55 aveva un “differentiated mechanism of action” e mostrava attività ansiolitica rispetto a THC. Non si tratta di conferma di beneficio clinico, ma mostra dove sta andando la chimica medicinale—lontano da una imitazione grossolana di THC e verso una farmacologia cannabinoid modellata sui target.[5]Psychoactive cannabinoid THC inhibits peripheral nociceptors by targeting NaV1.7 and NaV1.8 nociceptive sodium channels. Hebrew University of Jerusalem cannabinoids research portal. Research portal summary, 2025. https://cannabinoids.huji.ac.il/publications/psychoactive-cannabinoid-thc-inhibits-peripheral-nociceptors-targeting[6]A cannabis compound that relieves pain without the high. ScienceDaily. ScienceDaily, 2026. https://www.sciencedaily.com/releases/2026/06/260619033343.htm
Anche i canali del sodio meritano posto qui. Un report del 2025 della Hebrew University ha identificato l’inibizione da parte di THC dei nocicettori periferici tramite i canali del sodio nocicettivi NaV1.7 e NaV1.8. Si tratta di un risultato importante perché NaV1.7 e NaV1.8 sono target centrali del dolore, e il meccanismo è esterno a CB1/CB2. Si inserisce anche in una più ampia spinta traslazionale. Nel 2026, ScienceDaily ha evidenziato una ricerca su “a cannabis compound that relieves pain without the high”. Il composto esatto e le prospettive cliniche richiedono un esame accurato, ma la direzione è credibile: l’analgesia può, in linea di principio, essere separata dall’intossicazione centrale prendendo di mira vie periferiche o non CB1.
Bias di segnalazione Una proprietà di un ligando per cui stabilizza stati recettoriali che favoriscono una via a valle rispetto a un'altra, come la segnalazione della proteina G rispetto al reclutamento di beta-arrestin.
Affinità, efficacia, bias e finestre di concentrazione
Questa mappa dei target più ampia ha senso solo se i termini farmacologici sono chiari. Ki è una costante di affinità di legame: un Ki più basso di solito significa legame più stretto in un saggio di competizione. EC50 è la concentrazione che produce il 50 per cento di un effetto funzionale misurato. Non sono intercambiabili. Un ligando può legarsi strettamente ma produrre una segnalazione debole, oppure legarsi in modo modesto ma influenzare fortemente la funzione attraverso l’amplificazione di una via.
Un agonista attiva un recettore. Un antagonista blocca l’attivazione da parte di un altro ligando. Un agonista inverso spinge i recettori costitutivamente attivi verso una segnalazione basale più bassa. THC su CB1 è di solito descritto come un agonista parziale: anche quando occupa i recettori, non produce l’effetto pieno di un agonista ad alta efficacia. Questo aiuta a spiegare perché cannabinoid diversi, e persino diversi ligandi sintetici di CB1, possano avere tetti fisiologici molto differenti.
Il bias di segnalazione significa che un ligando stabilizza conformazioni del recettore che favoriscono una via rispetto a un’altra, per esempio segnalazione tramite proteina G rispetto al reclutamento di β-arrestina. Questo è ormai un pensiero standard nello sviluppo farmacologico, inclusa la chimica medicinale dei cannabinoid; l’articolo del 2016 sul Journal of Medicinal Chemistry “Library Docking for Cannabinoid-2 Receptor Ligands” si colloca in questa tradizione orientata al target. Desensibilizzazione significa che un’attivazione ripetuta o sostenuta può ridurre la responsività, un problema importante per i canali TRP e per CB1 stesso. Infine, l’impegno del target specifico per tessuto significa che lo stesso composto può colpire target diversi nel cervello, nell’intestino, nella pelle, nelle cellule immunitarie o nei nervi periferici a seconda di concentrazione, via di somministrazione, metabolismo ed espressione proteica locale. Ecco perché la promiscuità in vitro non equivale automaticamente a rilevanza clinica—ma anche perché le spiegazioni limitate a CB1/CB2 continuano a fallire.
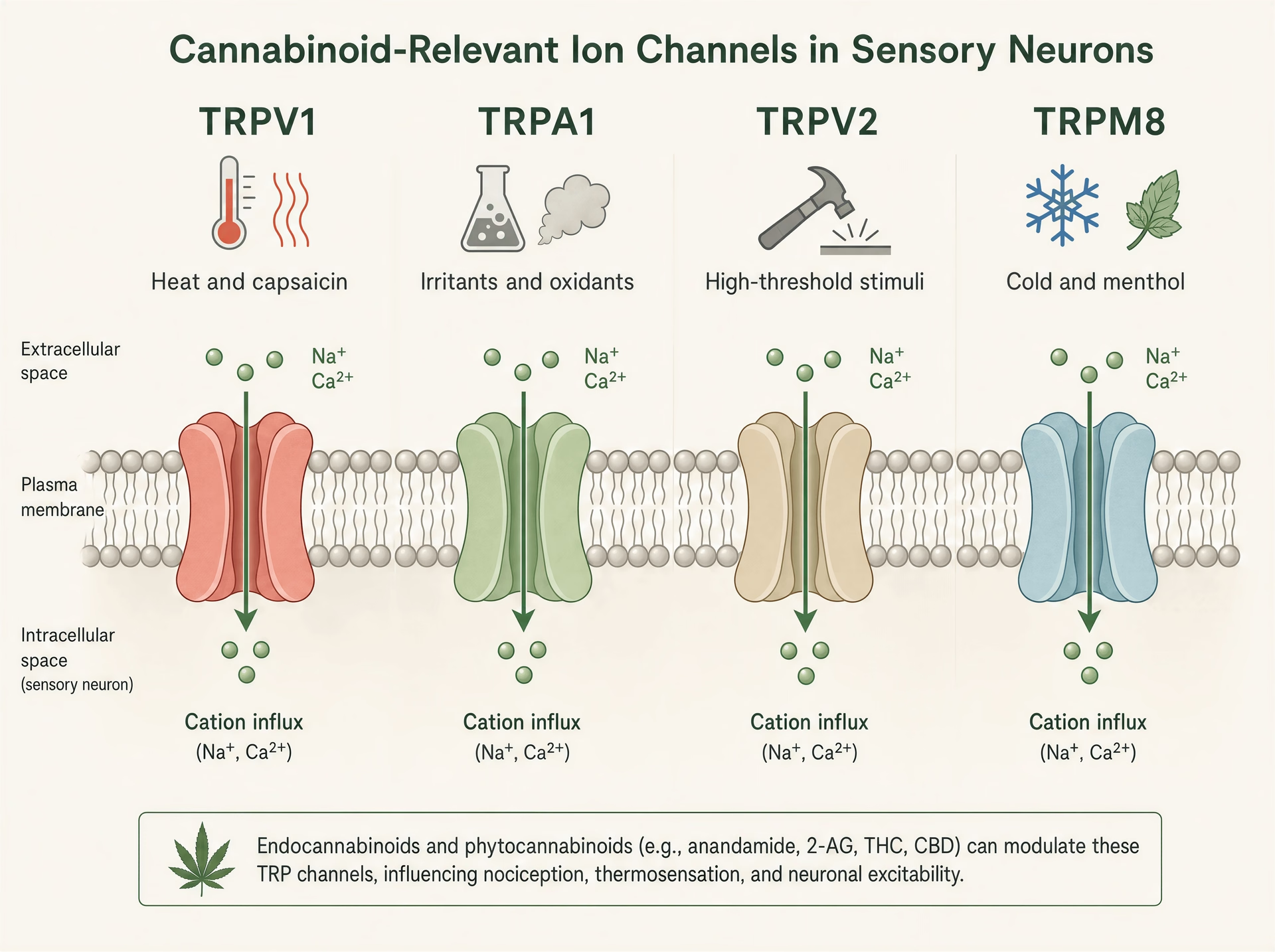
Canali TRP: i sensori di calore, dolore e irritazione su cui i cannabinoid continuano a intervenire
La solita formula sintetica dice che i cannabinoid agiscono attraverso CB1 e CB2. È troppo ristretta per spiegare ciò che molte di queste molecole fanno realmente nei tessuti. Ripetutamente, i fitocannabinoid colpiscono i canali recettoriali transitori, una superfamiglia di canali ionici presenti nei nocicettori, cheratinociti, nervi delle vie aeree, cellule immunitarie e altre interfacce sensoriali dove il corpo rileva calore, freddo, sostanze chimiche, stiramento, lesioni e infiammazione.[7]The Nobel Prize in Physiology or Medicine 2021. The Nobel Assembly at Karolinska Institutet. Nobel Prize Press Release, 2021. https://www.nobelprize.org/prizes/medicine/2021/press-release/
Questa biologia non è oscura. È stata centrale per la scienza somatosensoriale al punto che il premio Nobel per la Fisiologia o Medicina 2021 è andato a David Julius e Ardem Patapoutian “for their discoveries of receptors for temperature and touch.” Il lavoro di Julius nell’identificare il recettore della capsaicina, TRPV1, ha contribuito a stabilire la visione moderna secondo cui la segnalazione del dolore non è soltanto un filo che trasporta informazioni di danno; è chimicamente regolata fin dalla primissima terminazione sensoriale. Questo conta per i cannabinoid perché diversi cannabinoid vegetali interagiscono con la stessa dotazione molecolare che risponde al peperoncino, all’olio di senape, al calore nocivo, ad agenti rinfrescanti, a condizioni acide e ai lipidi infiammatori.
Il risultato è una farmacologia che appare confusa se ci si aspetta un solo recettore e un solo effetto. Ha più senso se si pensa in termini di controllo del guadagno sensoriale. Molti cannabinoid sono ligandi deboli o moderati dei recettori CB e, allo stesso tempo, modulatori diretti dei canali TRP. Alcuni li attivano. Alcuni li inibiscono. Alcuni fanno entrambe le cose a seconda di concentrazione, specie, variante di splicing, ambiente di membrana e del fatto che il saggio misuri ingresso di calcio, corrente, rilascio di neuropeptidi o comportamento in un animale.
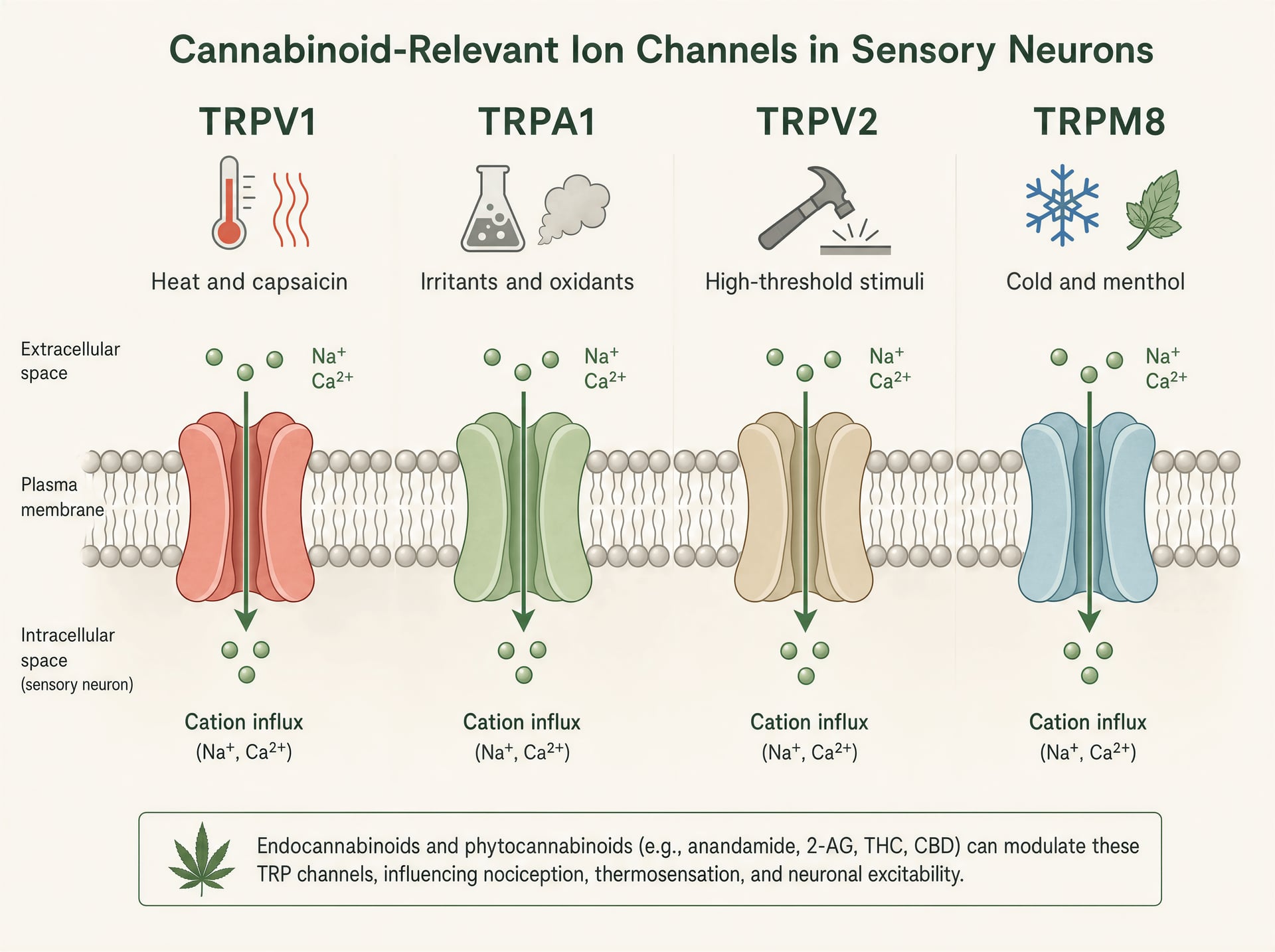
TRPV1, TRPA1, TRPV2 e TRPM8 nella biologia sensoriale
I canali TRP sono rilevatori polimodali. TRPV1 è il più noto: attivato da capsaicina, calore nocivo, protoni e mediatori infiammatori endogeni, è fortemente espresso nei neuroni sensoriali di piccolo diametro che guidano dolore urente e infiammazione neurogena. Aprendo il canale, i cationi entrano, depolarizzando il neurone e aumentando il calcio intracellulare. TRPA1 spesso si trova in popolazioni nocicettive sovrapposte ed è noto per rilevare irritanti elettrofili come l’isotiocianato di allile della senape e del wasabi, l’acroleina del fumo e i prodotti di stress ossidativo generati durante l’infiammazione. È rilevante non solo per il dolore ma anche per il prurito, la tosse, l’iperreattività delle vie aeree e la segnalazione trigeminale simile all’emicrania.
TRPV2 è meno lineare. In alcuni sistemi è un canale termico e meccanosensibile ad alta soglia, ma si trova anche in cellule immunitarie, glia e tessuti proliferativi, motivo per cui ricorre nelle discussioni su infiammazione e, più speculativamente, biologia del cancro. TRPM8, al contrario, è il sensore del freddo canonico attivato da temperature basse e da composti come mentolo e icilina. Eppure conta anche negli stati dolorosi, dove l’allodinia da freddo può diventare severa, e in alcuni contesti l’attività di TRPM8 può sopprimere il dolore attraverso una controstimolazione di circuito. Stessa famiglia, ruoli sensoriali molto diversi.
Questa ampiezza di funzioni spiega perché gli effetti dei cannabinoid possano apparire contraddittori in superficie. Attivare TRPV1 o TRPA1 può bruciare. Bloccare TRPM8 può ridurre le sensazioni di raffreddamento ma anche modificare il dolore da freddo. Stimolare TRPV2 in un tipo cellulare può influire sulla segnalazione del calcio senza produrre alcun effetto sensoriale evidente. Non esiste un singolo “effetto TRP” così come non esiste un singolo “effetto cannabinoid”.
CBD, CBG, CBC e THC sui canali della famiglia TRP
Tra i fitocannabinoid, CBD ha il profilo TRP più forte e replicato. In sistemi di espressione eterologa, CBD attiva i TRPV1, TRPA1 e TRPV2 umani a concentrazioni micromolari e inibisce TRPM8. Uno studio molto citato di De Petrocellis e colleghi del 2011, usando imaging del calcio in cellule HEK-293 trasfettate, ha trovato che CBD si comportava da agonista su TRPV1, TRPV2, TRPA1 e TRPV4, mentre antagonizzava TRPM8. La potenza non era uniforme: TRPA1 era particolarmente sensibile, con attività a bassa concentrazione micromolare, mentre altri canali richiedevano concentrazioni un po’ più alte. Questo schema si è confermato abbastanza da rendere il coinvolgimento dei TRP parte di ogni serio resoconto sulla farmacologia di CBD.
CBG e CBC si inseriscono nello stesso tema generale, pur con impronte proprie. CBG ha mostrato ripetutamente attività su TRPA1 e TRPV1, oltre a inibire TRPM8, rendendolo farmacologicamente interessante per modelli di dolore infiammatorio e ipersensibilità viscerale. CBC è meno studiato di CBD, ma i lavori in vitro disponibili suggeriscono che anch’esso attivi TRPA1 e possa coinvolgere TRPV1. Non si tratta di piccole curiosità trovate in un solo saggio e mai più osservate. Ricorrono in sistemi ricombinanti e in preparazioni sensoriali primarie, ed è proprio per questo che riemergono nei lavori sui meccanismi dell’analgesia e dell’infiammazione.
THC è più complicato. Può attivare TRPV2 ed è stato riportato che interagisca con TRPA1 e TRPV1 in alcune condizioni, ma in molti esperimenti la sua farmacologia è dominata dagli effetti mediati da CB1, soprattutto nel sistema nervoso centrale. Anche così, l’idea che THC agisca solo come farmaco CB1 è falsa. Un lavoro recente della Hebrew University, riportato nel 2025, ha sostenuto che THC inibisce i nocicettori periferici prendendo di mira i canali del sodio NaV1.7 e NaV1.8, un meccanismo non CB separato che si accorda con il punto più ampio qui: i cannabinoid spesso colpiscono più target rilevanti per il dolore allo stesso tempo. I canali TRP fanno parte di questa mappa non CB più ampia.
Serve cautela. Gran parte di queste evidenze proviene da saggi micromolari, e non tutti i micromolari in una piastra corrispondono a una concentrazione libera raggiungibile in un target umano. I cannabinoid lipofili si distribuiscono nelle membrane, legano proteine e generano metaboliti; la via di somministrazione e l’accumulo tissutale contano. Il fatto che la soluzione orale di CBD sia approvata dalla FDA per i disturbi convulsivi non dimostra che TRPV1 o TRPA1 siano i motori dei suoi effetti clinici nell’epilessia. Mostra soltanto che CBD fa chiaramente cose nell’uomo che non possono essere riassunte definendolo un “composto cannabinoid non intossicante”. La storia molecolare è più ampia di quell’etichetta.
L’attività sui TRP è anche sensibile al saggio. Un canale può apparire “attivato” in un saggio al calcio perché stano cambiando contemporaneamente riserve intracellulari, potenziale di membrana o lipidi endogeni. Le differenze di specie possono essere reali. Lo stesso vale per la dipendenza dallo stato. Il tessuto infiammato si acidifica, si ossida e produce mediatori lipidici, tutti elementi che rimodellano il gating dei TRP. Un cannabinoid che muove appena un canale a riposo può avere un effetto molto maggiore in un terminale nervoso lesionato.
Desensibilizzazione, analgesia e perché l’attivazione può ridurre il dolore
Questa è la parte che confonde i non specialisti: se TRPV1 e TRPA1 sono canali che producono dolore, perché mai la loro attivazione dovrebbe ridurre il dolore?
Perché l’attivazione acuta e l’output funzionale prolungato non sono la stessa cosa.
TRPV1 è l’esempio classico. La capsaicina brucia inizialmente, poi desensibilizza i nocicettori e può produrre analgesia dopo esposizione ripetuta o ad alta concentrazione. Clinicamente, questo principio è sfruttato nel cerotto di capsaicina all’8 per cento per il dolore neuropatico. Il meccanismo include desensibilizzazione calcio-dipendente, deplezione di neuropeptidi come sostanza P e CGRP, alterazioni dello stato di fosforilazione del canale e, in alcuni casi, defunzionalizzazione reversibile del terminale nervoso. Un canale che scarica intensamente all’inizio può diventare meno responsivo in seguito. Il segnale immediato è pronocicettivo; lo stato successivo può essere antinocicettivo.
I cannabinoid sembrano sfruttare la stessa logica. L’attivazione da parte di CBD di TRPV1 o TRPA1 può innescare l’ingresso di calcio, seguito da una ridotta responsività del canale e da una diminuzione dell’eccitabilità nei neuroni sensoriali. Questa è una delle vie plausibili attraverso cui un composto può bruciare in una piastra e tuttavia ridurre l’iperalgesia in un animale. L’asse temporale conta. Così come la dose. Concentrazioni basse possono sensibilizzare o attivare debolmente. Concentrazioni più alte possono indurre desensibilizzazione o persino effetti di membrana più ampi che sopprimono la scarica.
TRPA1 aggiunge un ulteriore livello perché è profondamente legato agli irritanti infiammatori e allo stress ossidativo. Nei sistemi delle vie aeree e trigeminali, l’attivazione ripetuta o prolungata può alterare il rilascio di neuropeptidi e la responsività riflessa. Questo lo rende rilevante per tosse, emicrania e stati di riacutizzazione infiammatoria, non solo per il “dolore” in senso ristretto. Se un cannabinoid coinvolge TRPA1 e poi riduce la responsività successiva, l’effetto netto può essere una minore segnalazione di irritazione anche se il primo evento molecolare è stato l’apertura del canale.
TRPM8 mostra spesso il pattern opposto nei saggi: cannabinoid come CBD e CBG spesso lo inibiscono anziché attivarlo. Questo potrebbe contare nell’ipersensibilità al freddo, dove una segnalazione eccessiva di TRPM8 contribuisce a un’allodinia dolorosa da freddo. Qui non c’è il paradosso dell’attivazione che porta sollievo; l’ipotesi più semplice è una soppressione diretta di una via di percezione del freddo. Ma anche questo non va sopravvalutato. In alcuni stati dolorosi, l’attività di TRPM8 può controbilanciare il dolore da caldo o il prurito, quindi bloccarlo non è automaticamente vantaggioso.
La posizione più forte supportata dalle evidenze è questa: i canali TRP non sono note marginali nella farmacologia dei cannabinoid. Sono target ricorrenti e funzionalmente rilevanti, soprattutto per effetti sensoriali periferici che coinvolgono calore, irritazione chimica, dolore infiammatorio, prurito e riflessi delle vie aeree. Non spiegano tutto. Non sono sempre il meccanismo dominante in vivo. Eppure chiunque cerchi di capire perché CBD, CBG, CBC o persino THC possano alterare dolore e infiammazione senza una corrispondenza netta con CB1 o CB2 deve avere TRPV1, TRPA1, TRPV2 e TRPM8 nella pagina fin dall’inizio, non come ripensamento.
Questo conta anche per il drug development. Le agenzie di salute pubblica stanno già distinguendo i cannabinoid familiari dagli intossicanti chimicamente alterati o potenziati perché le differenze a livello di target possono cambiare il rischio. Lo stesso principio si applica in senso inverso ai terapeutici: se l’analgesia può essere separata dall’intossicazione centrale, una strada è progettare composti che orientino l’azione verso i canali TRP periferici e altri target non CB invece che verso una forte agonismo di CB1 con penetrazione cerebrale. La vecchia storia riduzionista dei recettori è troppo piccola per i dati.
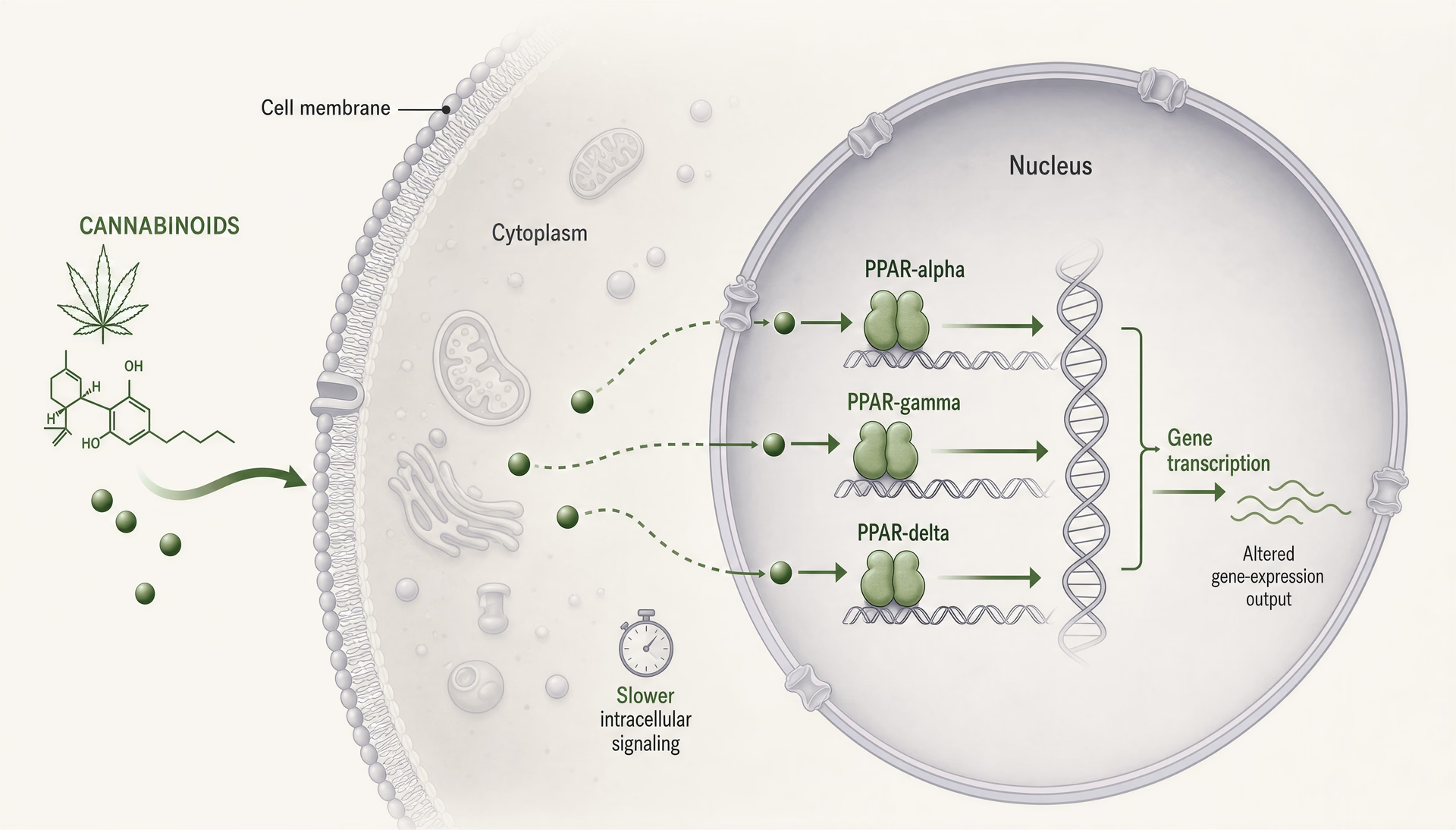
PPAR: i cannabinoid come segnali lipidici intracellulari, non solo come ligandi dei recettori di membrana
I recettori attivati da proliferatori dei perossisomi, di solito abbreviati in PPAR, cambiano il discorso sui cannabinoid perché si trovano in un luogo diverso e lavorano su un orologio diverso rispetto a CB1 e CB2. CB1 e CB2 sono recettori GPCR di membrana costruiti per una segnalazione rapida: secondi o minuti, canali ionici, rilascio di neurotrasmettitori, cascate chinasi. I PPAR sono recettori nucleari. Rispondono a molecole lipofile, mobilitano il macchinario trascrizionale e rimodellano quali geni una cellula esprime nell’arco di ore o giorni. Questo spostamento conta. Significa che alcuni effetti dei cannabinoid possono assomigliare meno all’agonismo recettoriale classico e più a una riprogrammazione del tono infiammatorio, del metabolismo mitocondriale, dell’ossidazione degli acidi grassi, della segnalazione fibrotica e delle risposte della glia.
Non è un allargamento speculativo. I cannabinoid sono altamente lipofili, si accumulano nelle membrane, si distribuiscono nei compartimenti intracellulari e generano metaboliti che possono avere profili di target diversi dalla molecola madre. Una classe di farmaci con queste proprietà è quasi progettata per incontrare i sensori lipidici nucleari. I PPAR sono tra i punti più plausibili in cui ciò avviene.
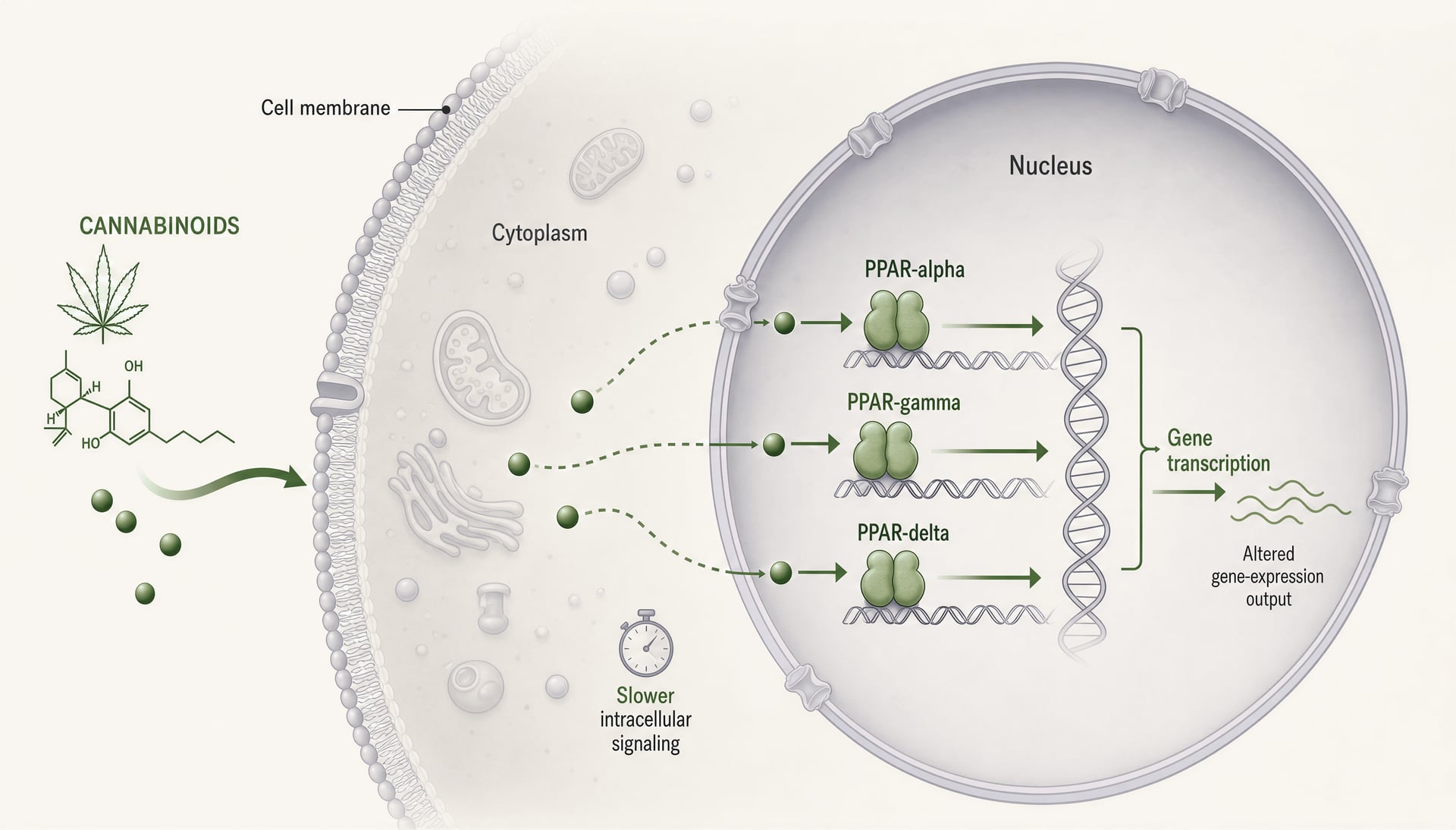
Cosa fanno PPAR-alpha, PPAR-gamma e PPAR-delta
Le tre principali isoforme dei PPAR si sovrappongono, ma non sono intercambiabili. PPAR-alpha è classicamente legato al catabolismo degli acidi grassi. È abbondante in fegato, cuore, rene, muscolo e altri tessuti che bruciano i grassi in modo intenso e, quando attivato, spinge programmi trascrizionali per beta-ossidazione, chetogenesi, gestione delle lipoproteine e ridotta segnalazione infiammatoria. I farmacologi lo conoscono dai farmaci fibrati. Nella ricerca sul dolore e sull’infiammazione, PPAR-alpha conta anche al di fuori del metabolismo perché può sopprimere l’espressione genica infiammatoria legata a NF-kappaB e alterare la segnalazione sensoriale.
PPAR-gamma è l’isoforma che continua ad apparire negli articoli sui cannabinoid, talvolta per buone ragioni e talvolta perché è il racconto più facile da raccontare. È molto rilevante per la differenziazione degli adipociti e la sensibilità all’insulina, ma questa scorciatoia lo sottovaluta. PPAR-gamma regola la polarizzazione dei macrofagi, la produzione di citochine, le risposte allo stress ossidativo, il rimodellamento fibrotico, il comportamento endoteliale e l’attivazione della glia nel sistema nervoso centrale. Questo gli conferisce un’evidente rilevanza per la malattia infiammatoria intestinale, la neuroinfiammazione, le complicanze diabetiche e la fibrosi tissutale. È anche un target a doppio taglio: una forte attivazione può migliorare la sensibilità all’insulina ma comportare edema, aumento di peso e altri rischi noti dai farmaci tiazolidinedionici.
PPAR-delta, detto anche PPAR-beta/delta, riceve meno attenzione nella divulgazione sui cannabinoid, ma non dovrebbe. È ampiamente espresso e sostiene l’uso degli acidi grassi, la funzione mitocondriale, la riparazione delle ferite, la biologia dei cheratinociti e alcuni programmi anti-infiammatori. A seconda del contesto, può frenare o favorire i processi patologici, motivo per cui la letteratura su di esso è meno ordinata. Se un cannabinoid o un metabolita di cannabinoid coinvolge PPAR-delta, l’esito biologico può variare per tessuto molto più di quanto suggerisca una storia semplice del tipo “agonista=beneficio”.
Dal punto di vista meccanicistico, tutte e tre le isoforme funzionano come fattori di trascrizione attivati da ligando che eterodimerizzano con il retinoid X receptor e si legano agli elementi di risposta ai proliferatori dei perossisomi nel DNA. Una volta attivati, non si limitano a premere un interruttore. Modificano reti trascrizionali. Co-attivatori, co-repressori, stato della cromatina, tipo cellulare, contesto infiammatorio e conformazione recettoriale specifica del ligando influenzano tutti il risultato. Due composti possono essere entrambi chiamati agonisti di PPAR-gamma e tuttavia guidare biologie significativamente diverse.
Questo punto è particolarmente importante per i cannabinoid, che sono spesso molecole farmacologicamente promiscue invece che strumenti puliti a target singolo.
CBD e cannabinoid correlati nella segnalazione metabolica e infiammatoria
CBD è l’esempio ricorrente perché il suo profilo clinico è mal spiegato solo da CB1 o CB2. La soluzione orale approvata dalla FDA per le crisi epilettiche nella sindrome di Lennox-Gastaut, nella sindrome di Dravet e nel complesso della sclerosi tuberosa mostra che CBD è farmacologicamente reale nell’uomo, ma non che un singolo target non cannabinoid spieghi le sue azioni. PPAR-gamma è uno dei candidati più citati perché numerosi studi cellulari e animali hanno collegato CBD a effetti anti-infiammatori e metabolici attenuati dagli antagonisti di PPAR-gamma o associati a cambiamenti trascrizionali dipendenti da PPAR-gamma.
Un articolo molto citato di O’Sullivan e colleghi del 2009 ha riportato che CBD causava vasorilassamento nelle arterie umane e che parte dell’effetto era sensibile all’antagonista di PPAR-gamma GW9662, suggerendo una componente dipendente da PPAR-gamma. Nel 2011, Esposito e coautori hanno mostrato in un modello cellulare simile all’Alzheimer che CBD riduceva la neuroinfiammazione indotta da beta-amiloide e che il blocco di PPAR-gamma riduceva questo effetto protettivo. Nel 2013, Hind e O’Sullivan hanno rivisto le evidenze secondo cui i cannabinoid possono attivare PPAR in modo diretto o indiretto, includendo CBD, THC, l’acido ajulemico, lipidi correlati all’anandamide e diversi cannabinoid sintetici.
Il pattern è abbastanza coerente da essere preso sul serio: CBD spesso si colloca in sistemi sperimentali dove i geni infiammatori diminuiscono, i marcatori di stress ossidativo calano e l’antagonismo di PPAR-gamma indebolisce la risposta. Ma prenderlo sul serio non equivale a considerarlo definito. Molti di questi studi usano concentrazioni micromolari di CBD. Questo conta perché le concentrazioni libere intracellulari nei tessuti umani vivi sono difficili da dedurre dalle concentrazioni nominali del bagno in una piastra. CBD inoltre si lega e altera le membrane, influisce sulla gestione del calcio, interagisce con i canali TRP, modula la segnalazione dell’adenosina inibendo il trasporto dei nucleosidi e può alterare il tono endocannabinoid. Ognuna di queste vie può alimentare cambiamenti trascrizionali che in seguito sembrano “simili a PPAR”.
I cannabinoid correlati aggiungono elementi al caso senza risolverlo. THC è stato segnalato in alcuni sistemi come attivatore di PPAR-gamma, anche se di solito debolmente rispetto ai ligandi dedicati. L’acido cannabidiolico e l’acido tetraidrocannabinolico hanno mostrato attività su PPAR in saggi selezionati. Lipidi correlati agli endocannabinoid come palmitoiletanolamide, oleoiletanolamide e alcuni derivati ossidati hanno relazioni più forti e consolidate con PPAR-alpha e PPAR-gamma rispetto ai fitocannabinoid più noti. Questa è una delle ragioni per cui il quadro del segnale lipidico intracellulare è migliore di un quadro ristretto del tipo “i cannabinoid vegetali si legano ai PPAR”. La specie attiva può essere il cannabinoid madre, un metabolita, un mediatore lipidico co-somministrato o uno spostamento a valle dei pool lipidici endogeni.
L’acido ajulemico è un caso di studio utile. È un analogo sintetico correlato a THC ma sviluppato intenzionalmente lontano dall’intossicazione classica. In diversi lavori preclinici ha mostrato azioni anti-infiammatorie e antifibrotiche con evidenze che implicano PPAR-gamma tra altri target. Questo tipo di chimica medicinale rispecchia una tendenza più ampia nel campo. Già nel 2016 un articolo dell’ACS Journal of Medicinal Chemistry intitolato “Library Docking for Cannabinoid-2 Receptor Ligands” rifletteva una progettazione basata sulla struttura invece di etichette recettoriali grossolane, e i nuovi programmi cannabinoid puntano sempre più a separare analgesia, ansioliticità o immunomodulazione dall’attivazione centrale di CB1. La stessa logica si applica agli scheletri attivi su PPAR: se una biologia cannabinoid utile può essere estratta tramite meccanismi trascrizionali e periferici, non c’è motivo per cui lo sviluppo del farmaco debba restare intrappolato nella farmacologia simile a THC.
I dati sulla segnalazione metabolica di CBD sono più misti di quelli anti-infiammatori. Alcuni studi preclinici suggeriscono migliore sensibilità all’insulina, riduzione delle adipochine infiammatorie o migliore gestione mitocondriale. Altri non mostrano benefici importanti e le evidenze nell’uomo sono scarse. La discussione pubblica qui corre spesso più veloce dei dati. Il fatto che PPAR-gamma controlli il glucosio e la biologia del tessuto adiposo non significa che CBD sia un modulatore metabolico clinicamente rilevante nell’uomo alle esposizioni standard.
Trascrizione genica, effetti ritardati e limiti delle prove
La biologia dei PPAR impone una correzione temporale. Se un effetto cannabinoid compare entro secondi o pochi minuti, i PPAR difficilmente ne sono la spiegazione primaria. La segnalazione dei recettori nucleari richiede in genere accesso del ligando ai compartimenti intracellulari, engagement del recettore, reclutamento diverso dei co-regolatori, cambiamenti trascrizionali e poi conseguenze a livello proteico. Questo richiede tempo. Ore sono plausibili. Giorni sono comuni. Quando gli articoli affermano che l’effetto rapido di un cannabinoid è “via PPAR-gamma”, lo scetticismo è appropriato a meno che il disegno separi chiaramente la segnalazione non genomica immediata dagli esiti trascrizionali successivi.
La progettazione del saggio è il problema ricorrente. I saggi reporter possono mostrare che un composto aumenta la trascrizione dipendente da PPAR, ma i sistemi reporter sono artificiali e possono amplificare un’attività debole. Gli studi con antagonisti sono informativi, ma farmaci come GW9662 non sono un siero della verità: gli effetti off-target e il blocco parziale complicano l’interpretazione. I saggi di legame aiutano, ma il legame diretto non garantisce che l’esposizione tissutale raggiunga la concentrazione necessaria in vivo. I modelli knockout sono più forti, anche se la compensazione da altre vie può offuscare i risultati. La migliore evidenza combina metodi: impegno diretto del target, farmacologia selettiva per il recettore, perturbazione genetica, concentrazioni tissutali rilevanti e una curva temporale coerente con l’azione trascrizionale. Molta della letteratura cannabinoid-PPAR non raggiunge questo standard.
La prominenza di PPAR-gamma nella ricerca su CBD è quindi sia giustificata sia esagerata. Giustificata, perché il segnale ricorre nei modelli vascolari, infiammatori, neurodegenerativi e fibrotici. Esagerata, perché CBD è esattamente il tipo di molecola lipofila e multi-target per cui concentrazione intracellulare, metaboliti attivi e contesto del saggio possono creare racconti meccanicistici seducenti ma incompleti. Una riduzione di TNF-alpha o IL-6 dopo esposizione a CBD non è un’impronta digitale. È un indizio.
Resta comunque valido il punto generale. I cannabinoid non dovrebbero essere considerati soltanto come ligandi dei recettori cannabinoid di membrana. Alcuni agiscono, direttamente o indirettamente, come segnali lipidici intracellulari che possono coinvolgere il macchinario trascrizionale nucleare. Questo apre vie plausibili a effetti anti-infiammatori, antifibrotici e neuroimmuni più lenti, meno legati all’intossicazione e potenzialmente più rilevanti per la modificazione delle malattie a lungo termine rispetto alla segnalazione acuta di CB1. Solleva anche una lezione regolatoria. Come le autorità hanno sottolineato in altri contesti, inclusa la dichiarazione HHS del 2025 secondo cui i prodotti potenziati con 7-hydroxymitragynine rappresentano “an imminent hazard to public safety,” le differenze a livello molecolare contano. Piccole modifiche strutturali possono dirottare l’impegno del target. Per i cannabinoid e i prodotti simili ai cannabinoid, ciò significa che la storia di sicurezza ed efficacia non può essere inferita solo dalla familiarità con THC, e la biologia dei PPAR è una delle ragioni.
GPR55, GPR18 e GPR119 e il problema dei GPCR orfani
Un GPCR orfano è un recettore accoppiato a proteine G il cui ligando endogeno, ruolo fisiologico o entrambi restano incerti. Un recettore deorphanizzato è uno per il quale è stato proposto e replicato abbastanza bene un attivatore endogeno convincente da sostenere una biologia operativa. Sembra ordinato. In pratica, raramente lo è. La farmacologia dei cannabinoid continua a scontrarsi con questo disordine perché endocannabinoid e fitocannabinoid sono lipofili, attivi sulla membrana e promiscui: possono modificare flusso di calcio, attività chinasica o trascrizione in modi che sembrano mediati da un recettore anche quando il target diretto è ancora controverso. È proprio così che GPR55, GPR18 e GPR119 sono entrati nella conversazione come “recettori cannabinoid non classici”.
La tentazione di coniare una nuova etichetta recettoriale è forte. Fa notizia. Ma supera le evidenze. GPR55 è arrivato più vicino a essere battezzato “CB3”, ma il campo non ha mai raggiunto la coerenza che ha sostenuto CB1 e CB2. La stessa cautela vale ancora di più per GPR18 e GPR119.
Perché GPR55 è stato una volta chiamato un possibile recettore cannabinoid
GPR55 è stato clonato nel 1999, e i primi studi di espressione lo collocavano in tessuti rilevanti per la biologia dei cannabinoid: regioni cerebrali, gangli della radice dorsale, milza, tratto gastrointestinale, vascolarizzazione, cellule immunitarie e cellule legate all’osso, inclusi osteoclasti e popolazioni della linea osteoblastica. Questa distribuzione contava. Un recettore espresso nelle vie del dolore, nei tessuti infiammatori e nell’osso invita subito al confronto con CB1 e CB2, soprattutto quando ligandi cannabinoid sembrano modificarne i readout.
Anche il suo profilo di segnalazione appariva abbastanza diverso da suscitare interesse. A differenza di CB1 e CB2, che si accoppiano principalmente a Gi/o e tendono a inibire l’adenilato ciclasi, GPR55 segnala più spesso tramite Gα12/13 e talvolta attraverso vie Gq-correlate, attivando RhoA, fosfolipasi C, ERK e rilascio di calcio intracellulare. Nei saggi cellulari, il readout caratteristico è spesso un transiente di calcio. Ciò rendeva GPR55 facile da “vedere” nei sistemi eterologhi, ma anche facile da sovrastimare, perché i saggi al calcio sono sensibili alla densità del recettore, al background cellulare, alla lipofilia del ligando e ai tempi del saggio.
Il motivo specifico per cui GPR55 è diventato candidato recettore cannabinoid era che diversi cannabinoid e ligandi simili ai cannabinoid producevano effetti misurabili su di esso. Ryberg e colleghi, scrivendo sul British Journal of Pharmacology nel 2007, hanno riportato che GPR55 poteva essere attivato da più ligandi cannabinoid e lo hanno proposto come “a novel cannabinoid receptor.” Quel lavoro è diventato la cerniera storica. Non ha risolto la questione; l’ha creata.
Subito dopo, sono emerse le crepe. Alcuni gruppi hanno trovato che il lisofosfatidilinositolo, in particolare specie 2-arachidonoyl LPI, era un agonista endogeno più convincente di qualunque cannabinoid classico. Oka e colleghi nel 2007 e lavori successivi hanno sostenuto con forza questa visione. Altri hanno osservato che i composti spesso discussi nella ricerca sui cannabinoid si comportavano in modo incoerente su GPR55: il cannabidiol (CBD) appariva spesso come antagonista o modulatore negativo in alcuni saggi, mentre Δ9-THC era debole, parziale o inattivo a seconda del sistema. Abnormal cannabidiol, O-1602 e alcuni cannabinoid sintetici mostravano talvolta attività più chiara di THC stesso. Non è ciò che ci si aspetta da un terzo recettore cannabinoid pulito.
Tuttavia, la biologia di GPR55 è reale, anche se l’etichetta è instabile. Nella ricerca sul dolore, il recettore è espresso nei neuroni sensoriali e nei circuiti spinali, e l’interruzione genetica o farmacologica della segnalazione di GPR55 ha ridotto l’ipersensibilità meccanica in alcuni modelli murini. Staton e colleghi in Pain (2008) hanno collegato l’attivazione di GPR55 al processamento del dolore infiammatorio e neuropatico, con antagonismo che riduceva l’ipersensibilità. Tuttavia, l’effetto non è universale tra modelli o ligandi. Alcuni dati suggeriscono una segnalazione pronocicettiva tramite mobilizzazione del calcio e aumento dell’eccitabilità neuronale; altri dataset sono più deboli o limitati al modello. La lettura più prudente è che GPR55 possa contribuire alla segnalazione del dolore in alcuni contesti, soprattutto infiammatori, ma non sia un interruttore maestro del dolore.
La biologia ossea offre un segnale più solido. Perché? Perché i fenotipi knockout di GPR55 sono più difficili da liquidare come artefatti di saggio. Nel 2009, Whyte e colleghi hanno riportato in PNAS che i topi privi di GPR55 mostravano maggiore massa ossea e funzione osteoclastica compromessa, sostenendo che GPR55 promuove il riassorbimento da parte degli osteoclasti. Questo aveva senso meccanicistico con la segnalazione legata a calcio e RhoA. Gli osteoclasti dipendono da rimodellamento del citoscheletro e gestione localizzata del calcio; GPR55 si adatta a quel macchinario meglio di CB1. Se un cannabinoid o un composto simile ai cannabinoid modula GPR55 qui, la conseguenza fisiologica potrebbe essere sostanziale.
L’infiammazione è il terzo grande tema. GPR55 è presente in cellule immuno-correlate e la sua attivazione è stata collegata al rilascio di citochine, al comportamento dei leucociti e a risposte infiammatorie vascolari. Ma anche qui la direzione non è perfettamente uniforme. In alcune preparazioni l’attivazione di GPR55 appare pro-infiammatoria, in altre più regolatoria, il che riflette probabilmente tipo cellulare, bias del ligando e cross-talk recettoriale piuttosto che una semplice contraddizione. Un recettore che si accoppia a più vie e si trova in ambienti di membrana diversi non produrrà un unico output universale.
Questa complessità spiega il lungo conflitto agonista/antagonista nella letteratura cannabinoid. CBD è l’esempio più chiaro. In diversi studi, CBD si è spesso comportato come antagonista o inibitore funzionale di GPR55, attenuando la segnalazione del calcio guidata da LPI. Lauckner et al. nel 2008, in un articolo molto citato su PNAS, hanno mostrato che l’attivazione di GPR55 aumentava il calcio intracellulare e promuoveva il rilascio di neurotrasmettitori, mentre CBD contrastava aspetti di quella segnalazione. Questo ha alimentato l’ipotesi persistente secondo cui alcuni effetti di CBD, specialmente nei modelli di crisi epilettiche e infiammazione, possano coinvolgere in parte il blocco di GPR55 anziché l’azione su CB1 o CB2. L’idea è plausibile. Non è dimostrato che sia il meccanismo dominante nell’uomo.
THC è ancora più ambiguo. Alcuni lavori lo classificano come agonista a bassa potenza di GPR55; altri trovano efficacia trascurabile; altri ancora suggeriscono un comportamento dipendente dalla riserva recettoriale o dalla via misurata. Un ligando può sembrare agonista in un saggio β-arrestina, neutro nel legame e antagonista in un saggio al calcio se il sistema è sovraespresso o biasato. Questo non è un dettaglio tecnico. È la storia.
Le prove miste su GPR18 e GPR119
GPR18 è stato spesso discusso perché in alcuni sistemi risponde a N-arachidonoyl glycine, un lipide correlato agli endocannabinoid, e perché abnormal cannabidiol e composti affini hanno mostrato effetti vascolari o immunitari che alcuni autori hanno mappato su GPR18. L’espressione è stata riportata in cellule immunitarie, microglia, milza e alcuni tessuti periferici. Ciò lo ha reso attraente come candidato per regolazione dell’infiammazione, traffico immunitario e forse dolore.
Ma la farmacologia è stata disomogenea fin dall’inizio. Kohno e colleghi nel 2006 hanno sostenuto l’attivazione di GPR18 da parte di N-arachidonoyl glycine. McHugh e colleghi hanno poi collegato GPR18 alla migrazione microgliale e alla segnalazione infiammatoria. Poi sono arrivati i problemi di replicazione. Alcuni laboratori non sono riusciti a riprodurre le risposte ai ligandi nei sistemi trasfettati. Altri hanno trovato una forte dipendenza dall’etichettatura del recettore, dalla linea cellulare o dall’ortologo di specie. Un recettore che “funziona” solo in un’architettura di saggio non è deorphanizzato in alcun senso stabile. Per i cannabinoid, l’evidenza è più debole di quanto suggeriscano i riassunti popolari. Potrebbe esserci una biologia reale, ma il caso per GPR18 come vero recettore cannabinoid resta debole.
GPR119 è diverso. È molto meno plausibile come recettore cannabinoid, nonostante la sua inclusione occasionale in elenchi ampi di recettori “non CB”. GPR119 è associato soprattutto al sensing lipidico nelle cellule beta pancreatiche e nelle cellule enteroendocrine, si accoppia a Gs per aumentare il cAMP e promuovere la secrezione di insulina dipendente dal glucosio e il rilascio di incretine. L’oleoiletanolamide è un candidato ligando endogeno meglio stabilito di qualunque cannabinoid classico. Poiché alcuni etanolamidi degli acidi grassi sono chimicamente adiacenti alla chimica degli endocannabinoid, GPR119 può essere trascinato nelle discussioni sui cannabinoid per associazione. Si tratta per lo più di confusione di categoria. La sovrapposizione è vicinanza chimica, non una forte evidenza che THC, CBD o i principali fitocannabinoid agiscano in modo significativo tramite GPR119 a concentrazioni fisiologiche.
Cosa sbaglia la farmacologia dei recettori orfani nei titoli
L’errore mediatico standard è semplice: un singolo saggio di segnalazione positivo diventa “gli scienziati hanno scoperto un nuovo recettore cannabinoid.” Quel salto ignora almeno quattro filtri.
Primo, la dipendenza dal saggio. Mobilizzazione del calcio, reclutamento di β-arrestina, fosforilazione di ERK, redistribuzione dinamica della massa e legame radioligand non fanno la stessa domanda. Un ligando lipofilo può perturbare membrane, alterare il traffico recettoriale o mostrare bias di via. Se il recettore è sovraespresso, i composti deboli iniziano a sembrare forti.
Secondo, le differenze di specie. GPR55 umano non è GPR55 murino in ogni dettaglio farmacologico, e lo stesso vale per GPR18. Un profilo del ligando costruito in cellule HEK293 con il recettore umano può non prevedere uno studio del dolore nel ratto.
Terzo, la concentrazione. Molti articoli sui cannabinoid riportano attività micromolare in vitro. Questo può contare farmacologicamente, ma non automaticamente. I livelli tissutali dopo inalazione, dosaggio orale, metabolismo di primo passaggio o accumulo locale nel grasso e nelle membrane variano enormemente. Il legame in vitro non è meccanismo clinico.
Quarto, il contesto. Un recettore nelle cellule immunitarie può mediare un effetto; lo stesso recettore negli osteoclasti, un altro. Aggiungendo il cross-talk con canali TRP, PPAR, recettori della serotonina e persino canali del sodio, la storia pulita di un ligando/un recettore si rompe rapidamente.
Ecco perché “CB3” non si è mai affermato. GPR55 ha una biologia credibile nella segnalazione del calcio, nel dolore, nel rimodellamento osseo e nell’infiammazione. Ha anche una farmacologia cannabinoid contraddittoria, forte sensibilità al saggio e una solida rivendicazione concorrente secondo cui i lipidi della famiglia LPI sono i suoi principali ligandi fisiologici. GPR18 è ancora più incerto. GPR119 appartiene per lo più a un altro insieme, se non come promemoria che i GPCR lipidici sono facili da associare in eccesso ai cannabinoid.
Per la scienza dei cannabinoid, la lezione è la moderazione. Questi recettori possono contare molto. Semplicemente non giustificano un rinominamento prematuro.
Segnalazione della serotonina: dove i cannabinoid si intersecano con i sistemi 5-HT
La serotonina è il punto in cui molte affermazioni popolari su CBD diventano sia più plausibili sia più scivolose. La parte plausibile è diretta: in saggi cellulari, modelli di ansia nei roditori, paradigmi di stress e un piccolo numero di studi sperimentali nell’uomo, 5-HT1A continua ad apparire come un nodo significativo negli effetti comportamentali di CBD. La parte scivolosa è che “agisce sulla serotonina” può significare diverse cose. Potrebbe significare agonismo diretto sul sito ortosterico. Potrebbe significare modulazione allosterica positiva. Potrebbe significare facilitazione della segnalazione del recettore senza legame ad alta affinità. Oppure potrebbe significare che CBD modifica l’attività di rete a monte o a valle dei neuroni serotoninergici, producendo un risultato dipendente dalla serotonina senza essere affatto un classico farmaco del recettore della serotonina.
Questa distinzione conta molto. Se un composto calma il comportamento in modo bloccato da un antagonista 5-HT1A come WAY-100635, ciò non prova da solo che il composto sia un agonista di 5-HT1A. Prova la dipendenza dalla segnalazione di 5-HT1A in quel modello. Non sono affermazioni identiche, e la copertura sui cannabinoid spesso le confonde.
5-HT1A e la questione dell’ansia
Il collegamento serotoninergico più forte per i cannabinoid, soprattutto CBD, è 5-HT1A. Questo recettore è un recettore serotoninergico accoppiato a Gi/o espresso sia come autorecettore sui neuroni serotoninergici del rafe sia come recettore postsinaptico in regioni rilevanti per l’ansia, tra cui ippocampo, amigdala e corteccia prefrontale. I farmaci che attivano o reclutano questo sistema possono ridurre l’ansia in alcuni contesti, ma la posizione del recettore conta: ridurre la scarica serotoninergica tramite autorecettori non è la stessa cosa che modellare la segnalazione postsinaptica nei circuiti limbici.
CBD è entrato in questa discussione attraverso lavori preclinici degli anni 2000 e 2010 che mostravano effetti ansiolitici in test come elevated plus maze, Vogel conflict test e paradigmi di paura contestuale, con blocco parziale da parte di WAY-100635. Un articolo molto citato è Campos e Guimarães, 2008, che ha trovato che CBD nel prelimbic riduceva le risposte cardiovascolari legate allo stress da contenimento e che i meccanismi 5-HT1A contribuivano all’effetto. Un altro studio umano importante è Bergamaschi et al., 2011: in un test di public speaking simulato, 600 mg di CBD orale riducevano l’ansia in soggetti con disturbo d’ansia sociale rispetto al placebo. Questo articolo non ha dimostrato la mediazione di 5-HT1A nell’uomo, ma si inseriva nel pattern preclinico e ha contribuito a rendere la serotonina un candidato meccanicistico serio anziché una frase di marketing.
La farmacologia recettoriale, però, non si è mai risolta in un semplice racconto “CBD è un agonista della serotonina”. I primi lavori in vitro suggerivano che CBD potesse dislocare i ligandi dai recettori umani 5-HT1A e comportarsi da agonista in alcuni saggi di segnalazione, ma le affinità erano modeste e dipendenti dal saggio. Russo e colleghi nel 2005 hanno riportato CBD come agonista dei recettori umani clonati 5-HT1A in saggi di legame [35S]GTPγS. Quel risultato fu influente, ma lavori successivi ne complicarono l’interpretazione. Alcuni gruppi vedevano un’attività diretta debole. Altri vedevano un potenziamento funzionale meglio spiegato da effetti allosterici o di membrana. La letteratura è coerente solo su un punto: 5-HT1A conta più per la farmacologia ansiolitica di CBD di quanto da soli possano spiegare CB1 o CB2.
Ecco perché il riduzionismo dei recettori fallisce. Se CBD fosse semplicemente un agonista pulito di 5-HT1A, il suo profilo dovrebbe assomigliare in modo più ordinato agli ansiolitici serotoninergici noti. Invece il segnale comportamentale è molto dipendente dal contesto, spesso con curve dose-risposta a U rovesciata. In alcuni test sui roditori, dosi moderate riducono il comportamento simile all’ansia mentre dosi più basse o più alte fanno meno. Questo è un segnale contro le narrazioni a un solo recettore. L’attivazione di TRPV1 a concentrazioni più alte è una ragione proposta. Lo sono anche gli effetti sul tono endocannabinoid, sul reuptake dell’adenosina e sulla gestione del calcio intracellulare. Una molecola può reclutare 5-HT1A e comunque rifiutarsi di comportarsi come un farmaco 5-HT1A da manuale.
Legame diretto versus effetti serotoninergici indiretti
Il modo migliore per leggere le evidenze sulla serotonina è per livelli. A livello molecolare, c’è supporto per un’interazione diretta tra CBD e 5-HT1A, ma non del tipo pulito, ad alta affinità e alta efficacia che chiude la questione. A seconda del sistema di saggio, CBD è stato descritto come agonista debole, agonista parziale o modulatore allosterico positivo. Il disaccordo non è semantica irrilevante. Gli agonisti ortosterici occupano il sito principale di legame della serotonina. I modulatori allosterici positivi cambiano il comportamento del recettore da un altro sito e possono amplificare le risposte della serotonina endogena senza attivare fortemente il recettore da soli. Questi meccanismi hanno implicazioni diverse per dose, tempi, effetti collaterali e traslazione nell’uomo.
I dati di segnalazione cellulare spesso indicano facilitazione più che attivazione bruta. In alcune preparazioni CBD potenzia le cascate di segnalazione mediate da 5-HT1A, compresi effetti su ERK e altre vie a valle, più di quanto la sua debole capacità di legame farebbe prevedere. Le spiegazioni possibili sono diverse. CBD è altamente lipofilo e si distribuisce nelle membrane, dove può alterare il microambiente del recettore e l’accoppiamento alla proteina G. Può anche aumentare indirettamente la segnalazione di anandamide, e il cross-talk endocannabinoid-serotonina nel rafe dorsale e nel prosencefalo è ben documentato. Poi c’è l’adenosina: CBD inibisce in alcuni sistemi l’attività del trasportatore dei nucleosidi equilibrativo, aumentando l’adenosina extracellulare e modificando l’eccitabilità neuronale in modi che possono alimentare i circuiti serotoninergici. Nulla di ciò rende 5-HT1A irrilevante. Lo rende integrato.
La farmacologia animale fornisce evidenze più forti della dipendenza dalla serotonina che dell’agonismo diretto. Ripetutamente, WAY-100635 attenua gli effetti di CBD in modelli di ansia, panico, nausea e stress. Resstel et al., 2009, per esempio, hanno collegato l’attenuazione delle risposte allo stress acuto da contenimento da parte di CBD a meccanismi 5-HT1A. Il lavoro di Rock e Parker sulla nausea e sulla nausea anticipatoria nei roditori ha implicato 5-HT1A nel profilo antiemetico di CBD. Si tratta di risultati utili, ma vanno letti come prove di via. Se bloccare 5-HT1A elimina l’effetto, la via è coinvolta. Non stabilisce se il recettore venga legato direttamente, modulato allostericamente o reclutato tramite cambiamenti di circuito.
Le prove nell’uomo restano modeste. Lo studio di Bergamaschi del 2011 è spesso citato perché ha mostrato un segnale ansiolitico misurabile nell’ansia sociale durante il public speaking. Studi di imaging più piccoli hanno riportato che CBD cambia l’attivazione limbica e paralimbica durante compiti di elaborazione emotiva. Tuttavia, nessuno di questi studi ha identificato l’occupazione del recettore 5-HT1A nell’uomo nel modo in cui studi PET possono farlo per farmaci serotoninergici consolidati. Questa assenza è importante. Stiamo inferendo il meccanismo dalla convergenza, non misurandolo direttamente a dosi cliniche.
Gli effetti calmanti del CBD dipendono in parte dalla segnalazione 5-HT1A.Limited evidence
Perché il profilo calmante di CBD resiste a etichette recettoriali semplici
CBD ha già un uso approvato dalla FDA, e non riguarda l’ansia. L’etichetta FDA 2024 per la soluzione orale di cannabidiol limita l’indicazione alle convulsioni associate alla sindrome di Lennox-Gastaut, alla sindrome di Dravet o al complesso della sclerosi tuberosa in pazienti di età pari o superiore a 1 anno. Questo dato è un utile controllo contro le esagerazioni. Un composto può avere segnali ansiolitici credibili senza avere un’efficacia nell’ansia definita al livello regolatorio, e può avere un coinvolgimento serotoninergico senza appartenere nettamente alla categoria dei farmaci serotoninergici.
Parte del problema è la scala. In vitro, i cannabinoid sono farmacologicamente confusi. In vivo, lo sono ancora di più perché distribuzione, metabolismo, accumulo tissutale e differenze di specie cambiano quali target contano. Un effetto su un recettore visto a 10 micromolare in cellule trasfettate può essere irrilevante dopo una normale dose orale, mentre un effetto apparentemente più debole in vitro può contare se il composto si concentra nel tessuto cerebrale ricco di lipidi o se contribuiscono metaboliti attivi. Questa è una delle ragioni per cui i titoli su “the serotonin receptor CBD hits” tendono a correre più veloci dei dati.
Un altro motivo è la biologia dei circuiti. L’ansia non è generata da un solo recettore. Emergere dall’interazione tra amigdala, bed nucleus of the stria terminalis, corteccia prefrontale mediale, ippocampo, ipotalamo e nuclei del tronco encefalico incluso il rafe dorsale. CBD sembra spostare l’attività in questa rete. Parte di questo spostamento probabilmente coinvolge 5-HT1A. Parte può coinvolgere TRPV1, che a dosi più alte può opporsi all’ansiolisi. Parte può dipendere da cambiamenti del tono di anandamide legati a FAAH, anche se l’inibizione di FAAH da parte di CBD alle esposizioni terapeutiche è discussa. Parte può riflettere effetti anti-infiammatori o autonomici che si ripercuotono sull’ansia percepita. Una volta adottata questa visione di rete, il fallimento di una spiegazione con una sola etichetta smette di sembrare una debolezza e inizia a sembrare un resoconto realistico della farmacologia.
Anche qui lo sviluppo farmacologico si sta muovendo. L’era della chimica medicinale è meno interessata a discutere se un composto sia “simile al THC” che a definire combinazioni di target e separare gli effetti desiderati dall’intossicazione. Questa logica appare in lavori ben lontani dalla serotonina, dalla selezione strutturale CB2 nell’articolo del 2016 sul Journal of Medicinal Chemistry “Library Docking for Cannabinoid-2 Receptor Ligands” fino ai più recenti tentativi di separare analgesia e compromissione centrale. Appare anche in programmi ansiolitici a livello aziendale. Nel 2025, MIRA Pharmaceuticals ha dichiarato che il proprio candidato MIRA-55 mostrava un “differentiated mechanism of action” e “anxiolytic activity relative to THC” in dati preclinici diffusi in un comunicato Nasdaq. Il livello di evidenza deve restare esplicito qui: preclinico, riportato dall’azienda, non prova clinica. Tuttavia, il segnale è significativo come indicatore di mercato e di ricerca. Le imprese stanno attivamente cercando agenti ispirati ai cannabinoid che possano calmare senza agire come THC, e i meccanismi rivolti alla serotonina fanno parte di questa ricerca.
Il contesto di salute pubblica rende la questione più che un dibattito accademico. Nel 2025, HHS ha affermato che 7-hydroxymitragynine “poses an imminent hazard to public safety” sostenendo un’azione di scheduling contro prodotti 7-OH potenziati pericolosi. Modifiche chimiche diverse creano profili di target e rischi diversi. La stessa lezione vale per lo spazio dei cannabinoid. Se un prodotto viene trattato come intercambiabile con i cannabinoid vegetali familiari solo perché suona vicino a THC o CBD, la farmacologia viene appiattita e la valutazione della sicurezza ne soffre.
Dove si colloca quindi l’evidenza? 5-HT1A è il meccanismo serotoninergico meglio supportato per gli effetti calmanti di CBD, ma l’affermazione più forte che i dati supportano oggi non è “CBD è un agonista della serotonina.” È più ristretta e difendibile: CBD produce spesso effetti ansiolitici e di buffering dello stress che dipendono in parte dalla segnalazione di 5-HT1A, mentre la modalità esatta di interazione sembra variare in base al saggio, alla dose, al tessuto e al contesto del circuito. Può sembrare meno ordinato di uno slogan a un recettore. È anche molto più vicino alla verità.
Oltre l’elenco richiesto: canali del sodio e altri target non canonici che stanno già cambiando il dibattito sul dolore
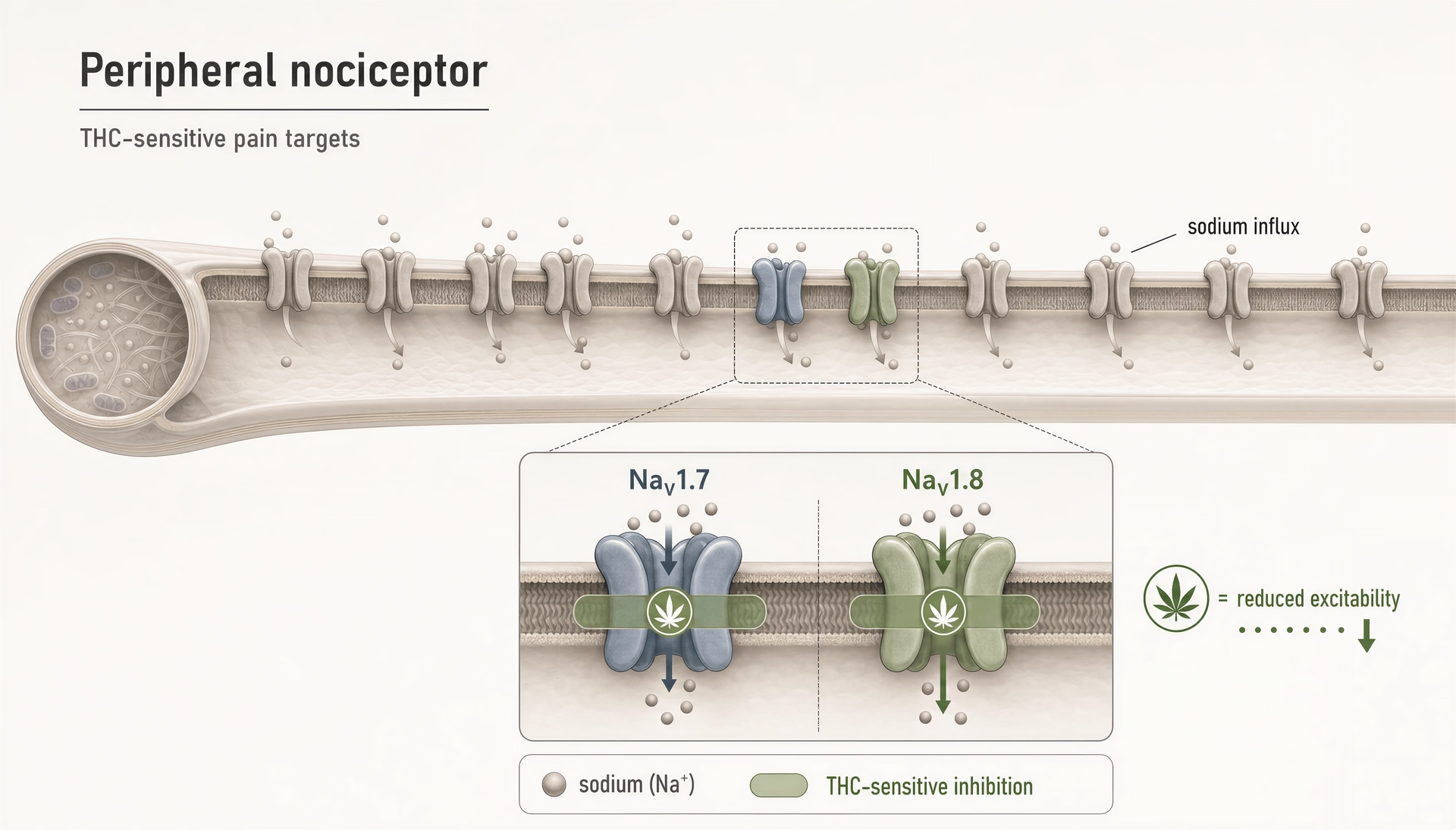
Per anni, la maggior parte delle discussioni pubbliche sulla farmacologia del dolore dei cannabinoid è rimasta intrappolata in una storia a due recettori: CB1 spiega gli effetti psicoattivi, CB2 spiega gli effetti immunitari e tutto il resto è secondario. Questo quadro è ormai troppo piccolo. Anche nel più ristretto campo del dolore, i cannabinoid non si limitano a toccare canali TRP, PPAR, GPCR orfani o vie legate alla serotonina. Interagiscono anche con canali del sodio voltaggio-dipendenti che si trovano al centro dell’eccitabilità dei nocicettori. È importante perché NaV1.7 e NaV1.8 non sono note marginali periferiche; sono tra le porte molecolari più studiate per la segnalazione del dolore nei neuroni sensoriali di piccolo diametro.
Il cambiamento non è soltanto accademico. I drug developer hanno trascorso anni cercando di bloccare la trasmissione del dolore a livello dei nervi periferici senza riprodurre sedazione, intossicazione, alterazioni della memoria e rischio di abuso associati a una forte attivazione centrale di CB1. Se un cannabinoid, o uno scheletro derivato da cannabinoid, può attenuare la scarica dei nocicettori agendo sui canali NaV al di fuori del cervello, si apre una logica terapeutica molto diversa. La conversazione si sposta da “Quanto fortemente colpisce CB1?” a “Dove agisce, a quale concentrazione e in quale tessuto?”
Questa mappa dei target più ampia si inserisce anche nel momento regolatorio più generale. Nel 2025, il Department of Health and Human Services degli Stati Uniti ha avvertito che “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety”, un promemoria che piccole modifiche chimiche possono produrre profili farmacologici e di sicurezza molto diversi. La politica sui cannabinoid ha spesso inseguito con ritardo questo fatto basilare. Trattare tutti i composti vicini agli intossicanti come se differissero solo per origine o per potenza equivalente a THC perde il punto. È la farmacologia a livello di target che predice effetto, rischio e potenziale come farmaco.
{kind=link}
{kind=link}
{kind=link}
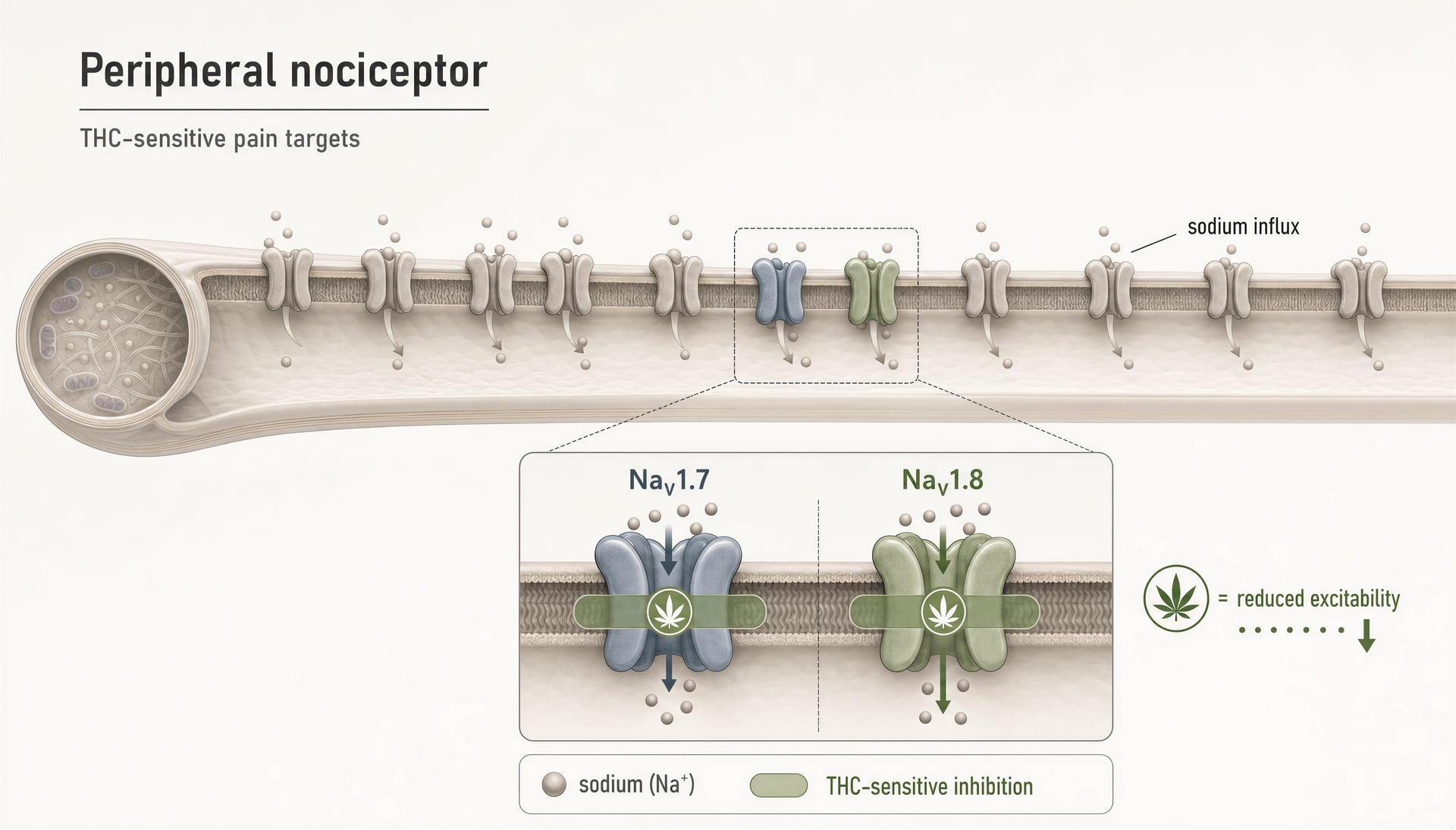
THC inibisce i nocicettori periferici prendendo di mira i canali del sodio NaV1.7 e NaV1.8.Preliminary evidence
THC sui canali nocicettivi periferici NaV1.7 e NaV1.8
La ragione più diretta per cui i canali del sodio appartengono ormai a qualunque mappa seria dei cannabinoid è il report del 2025 del gruppo della Hebrew University di Gerusalemme che mostra che THC inibisce i nocicettori periferici prendendo di mira i canali del sodio nocicettivi “NaV1.7 and NaV1.8.” Si tratta di un ampliamento significativo del vocabolario del campo. NaV1.7 e NaV1.8 sono fortemente espressi nei neuroni periferici sensibili al dolore, e il loro ruolo nella biologia del dolore umano non è speculativo. Le mutazioni loss-of-function di NaV1.7 possono produrre insensibilità congenita al dolore; le mutazioni gain-of-function possono guidare sindromi dolorose severe. NaV1.8 è altrettanto legato a stati di dolore infiammatorio e neuropatico perché sostiene la scarica ripetitiva nei nocicettori, soprattutto in condizioni depolarizzate.
Quindi, quando si dimostra che THC inibisce questi canali, il risultato non va nel cassetto degli “effetti off-target vari”. Indica un meccanismo che potrebbe ridurre direttamente l’eccitabilità delle fibre dolorose prima ancora che i segnali raggiungano il midollo spinale o il cervello.
È una classe meccanicistica diversa dalle storie cannabinoid più note. TRPV1, riconosciuto nel lavoro che ha contribuito alla quota di David Julius del premio Nobel 2021, può essere attivato o desensibilizzato da diversi cannabinoid, incluso CBD e CBG, con effetti che dipendono fortemente da dose e tempi. La segnalazione di PPAR-gamma è stata invocata per effetti anti-infiammatori e metabolici, spesso complicati dal fatto che l’accumulo intracellulare e i metaboliti possono contare quanto i composti madri. GPR55 resta abbastanza contestato da rendere ancora più slogan che scienza consolidata chiamarlo “CB3”. I collegamenti alla serotonina, soprattutto 5-HT1A, aiutano a spiegare parti del profilo ansiolitico di CBD, ma il circuito è dipendente dal contesto e spesso indiretto. L’inibizione dei canali del sodio è meno glamour. È anche, per il dolore, potenzialmente più pratica.
Un punto chiave è la promiscuità farmacologica. I cannabinoid sono spesso ligandi “sporchi” in senso tecnico: coinvolgono più target con affinità e conseguenze funzionali diverse. Questo non è un difetto della scienza; è la scienza. THC può ancora essere meglio noto per l’agonismo di CB1 nel SNC, ma ciò non annulla la capacità di modulare canali ionici periferici nelle condizioni giuste. La vera domanda è se tali condizioni siano raggiungibili in vivo in modi che aiutino più di quanto danneggino i pazienti. Il risultato della Hebrew University dice che questo è almeno plausibile al punto da meritare seria attenzione per lo sviluppo di farmaci.
Un analgesico cannabinoide può alleviare il dolore senza produrre lo sballo attraverso meccanismi periferici o non-CB1.Preliminary evidence
Analgesia periferica senza intossicazione centrale
È qui che il campo del dolore diventa interessante. Un meccanismo cannabinoid che riduce la scarica dei nocicettori periferici potrebbe, almeno in teoria, separare l’analgesia dalla compromissione cognitiva di solito legata all’attivazione di CB1 nel cervello. Questa distinzione è il centro del lavoro traslazionale attuale, non un vantaggio secondario.
Il report del 2026 di ScienceDaily ha espresso l’idea in linguaggio semplice: i ricercatori hanno identificato “a cannabis compound that relieves pain without the high.” Questa formulazione va letta con attenzione. È un segnale allo stadio di ricerca, non una terapia stabilita, e i riassunti popolari spesso comprimono i dettagli meccanicistici. Tuttavia, l’importanza traslazionale è evidente. Se l’attività analgesica può essere generata attraverso restrizione periferica, scarsa penetrazione cerebrale, impegno selettivo di target non CB1 o una combinazione dei tre, allora il vecchio compromesso tra sollievo dal dolore e intossicazione non è fissato dalla natura. È un problema di chimica medicinale.
Questo punto aiuta anche a spiegare perché il campo si sia mosso oltre le etichette recettoriali grossolane. L’articolo del 2016 sull’ACS Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands,” riflette un cambiamento più ampio verso la progettazione basata sulla struttura invece di trattare i cannabinoid come un’unica famiglia farmacologica con un solo asse utile di variazione. I chimici ora chiedono come modulare forma dello scheletro, lipofilia, bias recettoriale, distribuzione tissutale e destino metabolico. L’obiettivo non è solo l’attività più forte. L’obiettivo è un’attività selettiva nel posto giusto.
L’analgesia periferica è esattamente il tipo di endpoint in cui queste distinzioni contano. Un composto che attraversa male la barriera ematoencefalica ma inibisce in modo significativo NaV1.7 o NaV1.8 nei nocicettori potrebbe alleviare il dolore infiammatorio o neuropatico con molto meno intossicazione di THC stesso. Rimane un’ambizione, non un fatto clinico. Eppure il lavoro della Hebrew University fornisce a questa ambizione un appiglio molecolare.[8]Agriculture Improvement Act of 2018. U.S. Congress. Congress.gov, 2018. https://www.congress.gov/bill/115th-congress/house-bill/2/text
Rafforza anche il modo in cui pensiamo ai prodotti di cannabis già in circolazione. La definizione legale di hemp nel Farm Bill del 2018 si basa sul contenuto di delta-9 THC “not more than 0.3 percent on a dry weight basis.” Quel numero è regolatorio, non farmacologico. Non dice nulla sui canali del sodio, sull’attivazione dei TRP, sulla segnalazione di 5-HT1A, sui metaboliti attivi o sull’esposizione tissutale. Lo stesso vale per gli intossicanti più recenti potenziati o semi-sintetici. La sicurezza non può essere dedotta dalla storia di origine. Va dedotta dai target, dalle concentrazioni e dalla farmacocinetica reale.
Perché questi risultati contano per i futuri farmaci cannabinoid
La implicazione più forte della storia NaV1.7/NaV1.8 è che i futuri farmaci cannabinoid potrebbero avere successo proprio essendo meno “cannabinoid-like” nel senso popolare. Cioè, i discendenti utili della chimica del cannabis potrebbero non essere farmaci che imitano ampiamente il THC fumato. Potrebbero essere composti che prendono in prestito parte dello scheletro, evitano la segnalazione centrale di CB1 e agiscono invece su canali ionici periferici o su insiemi misti di target non canonici.
Questa possibilità si inserisce già nella più ampia base di evidenze. L’uso approvato di CBD riguarda i disturbi convulsivi, non l’analgesia di routine, e persino lì la farmacologia non è spiegata bene solo da CB1 o CB2. L’etichetta FDA della soluzione orale di cannabidiol afferma che è indicata per le convulsioni associate alla sindrome di Lennox-Gastaut, alla sindrome di Dravet o al complesso della sclerosi tuberosa in pazienti di 1 anno e oltre. In altre parole, l’unico grande farmaco cannabinoid approvato dalla FDA in uso diffuso resiste già al racconto semplificato dei recettori. Il campo del dolore sta raggiungendo la stessa lezione.
Anche i programmi aziendali iniziali puntano nella stessa direzione, pur richiedendo cautela. Nel 2025, MIRA Pharmaceuticals ha riportato dati preclinici affermando che MIRA-55 mostrava un “differentiated mechanism of action” e attività ansiolitica rispetto a THC. I comunicati aziendali non sono prove neutrali e i segnali preclinici spesso falliscono. Anche così, mostrano dove sta andando la chimica medicinale: lontano dall’imitazione non direzionale di THC e verso una progettazione modellata sul meccanismo.
Per il dolore, i canali del sodio possono diventare uno dei rami più importanti di questa strategia di progettazione. Non l’unico ramo. TRPV1, TRPA1, PPAR-gamma, GPR55, vie legate all’adenosina e modulazione serotoninergica resteranno presenti nel quadro. Ma NaV1.7 e NaV1.8 offrono qualcosa di particolarmente attraente: un collegamento diretto al comportamento elettrico delle fibre dolorose periferiche. Questo li rende target insolitamente concreti in un campo pieno di spiegazioni indirette.
Il risultato è un modo più pulito di pensare ai cannabinoid e al dolore. Non CB1 contro CB2. Non vegetale contro sintetico. E non “high” contro “medical” come se fossero categorie molecolari. La distinzione migliore è tra meccanismi di intossicazione centrale e meccanismi utili a livello periferico. Il risultato della Hebrew University colloca THC stesso su entrambi i lati di quella linea. È proprio per questo che conta.
| Cannabinoide | Bersagli non-CB1/CB2 enfatizzati nell'articolo | Principale cautela |
|---|---|---|
| CBD | TRPV1, TRPA1, TRPM8, 5-HT1A, PPAR-gamma, GPR55, segnalazione correlata all'adenosina | Molti segnali derivano da studi dipendenti dal saggio e spesso a scala micromolare |
| THC | TRPV2, interazioni riportate con TRPV1/TRPA1, NaV1.7, NaV1.8 | Gli effetti centrali di CB1 possono predominare e oscurare altri meccanismi |
| CBG | TRPA1, TRPV1, TRPM8, interazioni alpha-2 adrenergiche e correlate a 5-HT1A | La traduzione dalla concentrazione in vitro all'esposizione umana è incerta |
| CBC | TRPA1, canali della famiglia TRPV | Le evidenze nell'uomo sono scarse |
| THCV | Possibilità non-CB1 incluse segnalazione TRP e metabolica | Dose e contesto possono cambiare il comportamento apparente |
| CBDA / THCA | Azioni correlate a 5-HT1A, TRP ed enzimi discusse nel lavoro preclinico | Stabilità, decarbossilazione ed esposizione complicano l'interpretazione |
In cosa differiscono i cannabinoid specifici quando si smette di chiedere solo di CB1 e CB2
Una volta che si smette di trattare CB1 e CB2 come l’intera storia, l’elenco familiare dei cannabinoid appare molto meno ordinato. Queste molecole non sono chiavi pulite per due serrature. Sono composti lipofili, sensibili alla concentrazione, che possono colpire canali ionici, recettori nucleari, processi di trasporto, GPCR orfani e, in alcuni casi, canali del sodio voltaggio-dipendenti. Questo conta perché dolore, infiammazione, controllo delle crisi epilettiche, ansia ed effetti avversi spesso si mappano male su etichette semplici come “agonista di CB1” o “agonista di CB2”.
CBD ha forzato più di ogni altro cannabinoid questo cambiamento. La sua debole efficacia sui recettori cannabinoid classici ha reso il vecchio quadro difficile da difendere, soprattutto quando una soluzione orale di cannabidiol approvata dalla FDA ha ottenuto indicazioni per convulsioni associate alla sindrome di Lennox-Gastaut, alla sindrome di Dravet e al complesso della sclerosi tuberosa in pazienti di età pari o superiore a 1 anno. Un cannabinoid clinicamente utile con una intossicazione limitata simile a CB1 era un problema per il riduzionismo recettoriale. I ricercatori hanno dovuto guardare altrove.
Il campo più ampio ha seguito. Il lavoro sui canali TRP si è appoggiato alla biologia sensoriale che è stata riconosciuta al massimo livello quando il premio Nobel per la Fisiologia o Medicina 2021 è andato a David Julius e Ardem Patapoutian per le scoperte dei recettori per temperatura e tatto. La farmacologia dei cannabinoid interseca direttamente questa biologia: TRPV1, TRPA1 e TRPM8 ricorrono continuamente perché molti fitocannabinoid possono attivarli, inibirli o desensibilizzarli a concentrazioni sperimentalmente rilevanti. Questo non significa che ogni risultato di saggio predica un effetto nell’uomo. Significa che la vecchia idea secondo cui la “vera” azione cannabinoid inizia e finisce su CB1/CB2 è errata.
CBD: il prototipo della complessità non CB1/CB2
CBD è il miglior esempio di perché la promiscuità di target conti. Ha bassa affinità ed efficacia limitata su CB1 e CB2 rispetto a THC, ma chiaramente fa qualcosa di biologicamente importante. Il divario tra questi due fatti ha prodotto una serie di ipotesi di lavoro, alcune più solide di altre.
I canali TRP sono stati tra le prime alternative serie. CBD attiva TRPV1 nei sistemi eterologhi, e TRPV1 non è un sentiero laterale oscuro; è un canale centrale per nocicezione e dolore infiammatorio. L’attivazione può sembrare controintuitiva se l’obiettivo terapeutico è l’analgesia, ma l’attivazione ripetuta o sostenuta di TRPV1 spesso produce desensibilizzazione, riducendo l’eccitabilità successiva. Questa è una ragione per cui la farmacologia TRP può sembrare contraddittoria sulla carta. Un composto può attivare prima e quietare il sistema dopo. CBD mostra anche azioni su TRPA1 e può inibire TRPM8 in alcuni modelli, rendendolo farmacologicamente più ampio della consueta sintesi pubblica di “cannabinoid non intossicante”.
La storia della serotonina è più contestata ma resta importante. Una vasta letteratura preclinica collega CBD a effetti correlati a 5-HT1A, soprattutto in modelli di ansia, stress e nausea. L’affermazione più pulita non è che CBD sia un semplice agonista ad alta affinità di 5-HT1A come si parla della farmacologia legata a buspirone, ma che la segnalazione di 5-HT1A contribuisce spesso agli effetti di CBD in vivo, talvolta attraverso agonismo parziale, talvolta tramite meccanismi allosterici o di circuito ancora non definiti. Questa distinzione conta. Troppe sintesi appiattiscono “coinvolge 5-HT1A” in “funziona tramite serotonina.” I dati non supportano tale semplificazione.
PPAR-gamma è un altro candidato ricorrente, e qui la chimica intracellulare conta. I PPAR sono recettori nucleari, quindi una molecola lipofila che si distribuisce in membrane e cellule può influenzarli in modi che un modello basato solo su recettori di superficie non coglie. CBD è stato riportato come attivatore di PPAR-gamma in sistemi cellulari, e la segnalazione PPAR-gamma ha legami plausibili con infiammazione, metabolismo lipidico, fibrosi e neuroinfiammazione. Ma c’è un problema: alcuni effetti legati a PPAR possono riflettere metaboliti, esposizione prolungata o cambiamenti indiretti nei mediatori lipidici endogeni piuttosto che un’occupazione recettoriale in un solo passaggio. La farmacologia è abbastanza reale da meritare studio serio, ma meno adatta a diventare slogan.
La segnalazione dell’adenosina è entrata nel quadro perché CBD sembra inibire in alcuni sistemi il trasporto equilibrativo dei nucleosidi, potenzialmente aumentando il tono extracellulare dell’adenosina e influenzando indirettamente le vie anti-infiammatorie legate ad A2A. Anche questo non è una farmacologia di legame pulita a un recettore. È farmacologia del trasporto e del contesto tissutale. Se ciò suona più disordinato di “CBD colpisce CB2”, lo è. È anche più plausibile.
Poi c’è GPR55, spesso proposto come un ipotetico “CB3.” Questa etichetta è ancora troppo sicura. CBD può antagonizzare o comunque modulare la segnalazione legata a GPR55 in diversi sistemi sperimentali, e GPR55 è stato implicato in eccitabilità, biologia ossea, infiammazione e vie correlate al cancro. Ma i ligandi endogeni del recettore, il coupling di via e la rilevanza traslazionale restano dibattuti. GPR55 è un utile generatore di ipotesi, non un sostituto definitivo di CB1/CB2.
La conclusione è semplice: CBD è diventato il prototipo della farmacologia cannabinoid non CB1/CB2 perché i suoi effetti clinicamente rilevanti non potevano essere spiegati altrimenti. Questo è ancora vero.
CBG, CBC, THCV, cannabinoid acidi e la farmacologia dei minor cannabinoid
I minor cannabinoid sono spesso venduti al pubblico come se ciascuno avesse un tratto caratteriale singolo. La farmacologia non collabora.
CBG viene di solito descritto come lievemente attivo su CB1 e CB2, ma i segnali più interessanti si trovano fuori da questi recettori. In alcuni saggi interagisce con i sistemi adrenergici alfa-2 e 5-HT1A, mostra attività sui canali TRP ed è stato studiato per effetti anti-infiammatori e analgesici che non si riducono facilmente all’attivazione classica dei recettori cannabinoid. Illustra anche un problema ricorrente: l’attività micromolare in vitro è facile da pubblicare e difficile da tradurre. Un colpo recettoriale a 10 micromolare può contare in una piastra ma non nel plasma umano, oppure può contare solo nei tessuti in cui il composto si concentra.
CBC è rimasto a lungo meno caratterizzato rispetto all’hype che lo circonda. Sembra coinvolgere in modo più convincente i canali TRPA1 e della famiglia TRPV che CB1, e alcuni studi suggeriscono effetti anti-infiammatori o simili all’analgesia negli animali. C’è anche interesse per l’interazione di CBC con il tono endocannabinoid, compresi effetti che potrebbero alterare indirettamente la segnalazione dell’anandamide. Ma “CBC agisce attraverso i canali TRP” è ancora un punto di partenza, non una risposta definitiva. Le evidenze umane sono molto scarse.
THCV è un caso più complicato perché può comportarsi in modo diverso in base alla dose e al contesto anche su CB1, spesso descritto come antagonista neutro o ligando a bassa efficacia a certe concentrazioni e composto più simile a un agonista in altre. Al di fuori di CB1/CB2, THCV è stato collegato ad azioni sui TRP e a effetti metabolici, con interesse ripetuto per appetito, controllo glicemico ed equilibrio energetico. Parte di quell’entusiasmo ha superato le evidenze anni fa. L’interpretazione migliore è che THCV sia farmacologicamente interessante proprio perché non si adatta al modello di THC, non perché un singolo target secondario abbia spiegato pienamente il suo profilo.
I cannabinoid acidi meritano più attenzione di quanta ne ricevano. THCA e CBDA sono spesso trattati semplicemente come precursori “grezzi”, ma la loro farmacologia non è solo quella di composti inattivi in attesa della decarbossilazione. CBDA ha alcune delle evidenze più interessanti per effetti anti-nausea legati a 5-HT1A nei lavori preclinici, e sia i cannabinoid acidi sia altri composti hanno mostrato azioni sui TRP e sugli enzimi in vitro. La loro minore penetrazione cerebrale rispetto ai cannabinoid neutri può persino essere un vantaggio per alcuni target periferici o gastrointestinali. Il problema è la qualità dei dati e il realismo della dose. Molte affermazioni si basano su studi animali scarsi o saggi cellulari con rilevanza umana incerta.
Qui la politica attuale e la chimica del prodotto si scontrano con la farmacologia. Il Farm Bill del 2018 ha definito l’hemp usando una soglia di delta-9 THC di non più dello 0,3 per cento sul peso secco. Quel confine legale non dice nulla su GPR55, TRPV1, NaV1.7, metaboliti o analoghi semi-sintetici. I regolatori hanno iniziato a reagire a questa discrepanza in aree affini. Nel 2025, HHS ha affermato che “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” sostenendo un’azione della DEA sui prodotti 7-OH potenziati. Classe di farmaco diversa, stessa lezione: prodotti modificati o arricchiti possono avere profili di sicurezza che divergono nettamente dal materiale di partenza. I cannabinoid non sono esenti da questa logica.
Metaboliti, affermazioni sull’entourage effect e il problema chimico
Una delle ragioni per cui la farmacologia dei cannabinoid diventa scivolosa è che il composto madre non è sempre l’intera esposizione. I metaboliti possono contare, a volte molto. Il pubblico lo ha appreso soprattutto attraverso i metaboliti intossicanti di THC, ma il principio più ampio si applica a tutta la scienza dei cannabinoid. Via di somministrazione, metabolismo di primo passaggio, partizione tissutale e differenze di specie possono tutti cambiare quali target vengano realmente coinvolti.
Anche THC, il composto più strettamente associato a CB1, non è limitato alla biologia CB1. Nel 2025, ricercatori della Hebrew University hanno riportato che THC inibisce i nocicettori periferici prendendo di mira i canali del sodio nocicettivi NaV1.7 e NaV1.8. Questo ricorda in modo importante che l’analgesia può coinvolgere effetti diretti sul macchinario dell’eccitabilità, non solo segnalazione GPCR. Un report di ricerca del 2026 evidenziato da ScienceDaily ha spinto la stessa idea traslazionale più avanti, descrivendo un composto derivato dal cannabis che alleviava il dolore senza il high. Questo resta un’evidenza allo stadio di ricerca, non medicina consolidata, ma indica una seria strategia di drug development: separare analgesia dall’intossicazione centrale sfruttando target non CB1, restrizione periferica o entrambi.
La chimica medicinale si sta muovendo in quella direzione. La selezione basata sulla struttura attorno ai recettori cannabinoid era già ben avviata con l’articolo del 2016 sul Journal of Medicinal Chemistry “Library Docking for Cannabinoid-2 Receptor Ligands”, e i nuovi sforzi puntano sempre più a meccanismi differenziati invece che a composti genericamente “THC-like ma più deboli”. Un comunicato del 2025 di MIRA Pharmaceuticals, che va trattato come dato preclinico riportato dall’azienda e non come conferma indipendente, descriveva MIRA-55 come avente un meccanismo differenziato e attività ansiolitica rispetto a THC. Il punto importante non è che l’affermazione sia provata. È che gli sviluppatori di farmaci ora presumono che la separazione dei target conti.
Qui entra in gioco anche l’entourage effect. Come idea scientifica ristretta, è plausibile che combinazioni di cannabinoid, terpeni e metaboliti possano spostare la farmacocinetica o produrre effetti additivi, opposti o talvolta supra-additivi su più target. Come spiegazione onnicomprensiva del perché un prodotto di cannabis misto funzionerebbe sempre meglio, è di solito troppo vaga per essere testata e troppo elastica per essere falsificata.
Il problema chimico è elementare. Una miscela contenente THC, CBD, CBG, cannabinoid acidi, prodotti ossidati, terpeni residui e metaboliti variabili non è un’unica intervento. Sono molti componenti in movimento i cui effetti dipendono dai rapporti di concentrazione e dal timing. Esiste una farmacologia multi-target plausibile. Esiste anche una semplificazione non supportata. Non sono la stessa cosa.
Lo standard migliore è l’evidenza specifica per target collegata all’esposizione. Quale composto, a quale concentrazione, in quale tessuto, producendo quale effetto misurabile? Una volta poste queste domande, la mitologia svanisce e la vera storia dei cannabinoid emerge: non due recettori, ma una mappa farmacologica affollata.
I metodi contano: perché la progettazione dei saggi modella ciò che pensiamo facciano i cannabinoid
Le affermazioni sui target dei cannabinoid spesso sembrano più pulite dei dati che le sostengono. Un articolo dice che CBD “attiva TRPV1”, un altro che è un “agonista di 5-HT1A”, un terzo definisce GPR55 un candidato recettore cannabinoid e uno studio di docking propone una posa ordinata in CB2 o in qualche tasca di GPCR orfano. Queste affermazioni possono essere utili in senso direzionale. Non sono tutte lo stesso tipo di prova.
Questa distinzione conta perché i cannabinoid sono molecole grasse, amanti delle membrane, con la cattiva abitudine di sembrare attive in più luoghi di quelli che davvero contano in vivo. Il campo l’ha imparato a caro prezzo. Se un composto si partiziona nelle membrane, cambia le proprietà del doppio strato, si accumula dentro le cellule, forma metaboliti attivi o mostra un effetto solo a 10-50 micromolare, può generare narrazioni di target che crollano quando vengono testate con condizioni più rigorose. Per la farmacologia del cannabis, i metodi non sono un dettaglio tecnico. Decidono quali meccanismi sopravvivono.
Saggi di legame, saggi funzionali e studi di docking
Partiamo dalla domanda più antica e pulita: il composto si lega? In un saggio di legame radioligando, membrane o cellule intatte che esprimono un recettore vengono incubate con un ligando radioattivo noto più concentrazioni crescenti del composto in prova. Se la molecola di prova spiazza il radioligando, gli investigatori stimano l’affinità, spesso riportata come Ki. È utile. È anche limitato. Il legame dice che una molecola può occupare un sito nelle condizioni del saggio; non dice cosa succede dopo.
Ecco perché i saggi funzionali contano più del legame quando si fanno affermazioni sulla segnalazione. Per i canali TRP come TRPV1, TRPA1 o TRPM8, i ricercatori usano spesso saggi di flusso di calcio con coloranti fluorescenti. Se l’apertura del canale fa entrare calcio nella cellula, la fluorescenza aumenta. Il vantaggio è ovvio: questi saggi si scalano bene e possono confrontare rapidamente molti composti. Il problema è altrettanto ovvio se si conoscono i cannabinoid. Alcuni cannabinoid sono fluorescenti, alcuni perturbano le membrane, alcuni liberano calcio indirettamente da riserve intracellulari e alcuni scatenano desensibilizzazione del canale dopo un’attivazione iniziale. Un singolo picco in una traccia di calcio può nascondere diversi meccanismi.
Il patch clamp è più lento ma molto più informativo. Registra direttamente la corrente ionica. Per i canali ionici, specialmente i canali del sodio come NaV1.7 e NaV1.8, il patch clamp può mostrare se il farmaco modifica attivazione, inattivazione, probabilità di apertura o densità di corrente. Ecco perché il report della Hebrew University del 2025 su THC che agisce nei nocicettori periferici attraverso NaV1.7 e NaV1.8 è metodologicamente importante: l’elettrofisiologia diretta può separare un vero effetto sul canale da un vago segnale cellulare. Se un cannabinoid riduce la corrente del sodio nei nocicettori a concentrazioni ottenute nel tessuto periferico, ciò ha più peso meccanicistico per l’analgesia di un altro vago “interazione recettoriale” come etichetta.
GPCR e recettori nucleari richiedono letture diverse. Per i GPCR, gli investigatori possono misurare cAMP, reclutamento di beta-arrestina, legame GTPγS, fosforilazione di ERK o segnali di calcio in linee cellulari ingegnerizzate. Questi non sono intercambiabili. Un cannabinoid può sembrare agonista in una via, agonista parziale in un’altra e antagonista in una terza. Non è sciatteria; è bias di segnalazione. Per PPAR-gamma, l’approccio comune è un saggio reporter in cui un promotore sensibile a PPAR guida la luciferasi. La luce aumenta e il composto viene chiamato attivatore di PPAR. Ma i saggi reporter sono diversi passaggi lontani dall’impegno diretto del target. Un cannabinoid lipofilo può alterare la trascrizione indirettamente attraverso stress cellulare, metabolismo o cambiamenti nei mediatori lipidici endogeni.
Le letture dell’espressione genica sono ancora più a valle. Se un articolo mostra che CBD modifica i trascritti infiammatori e che l’effetto si riduce con un antagonista di PPAR, questo è suggestivo, non definitivo. L’antagonista può essere imperfetto. Le cellule possono produrre metaboliti. Il cannabinoid potrebbe alterare il tono dell’adenosina, la gestione del calcio, lo stato redox o l’ordine della membrana. Il meccanismo per sottrazione è fragile.
Poi c’è il docking. Il docking chiede se una piccola molecola possa entrare in una tasca di legame modellata o determinata sperimentalmente e ottenere un punteggio favorevole. Se usato bene, è triage di chimica medicinale. Se usato male, diventa certezza decorativa. L’articolo del 2016 dell’ACS Journal of Medicinal Chemistry, “Library Docking for Cannabinoid-2 Receptor Ligands”, è un buon punto di accesso perché mostra la logica nella sua forma migliore: lo screening basato sulla struttura può aiutare a prioritizzare gli scaffold, identificare contatti plausibili ligando-recettore e guidare la sintesi attorno alla selettività per CB2. Questo è ciò a cui serve il docking. Genera ipotesi. Non dimostra che un fitocannabinoid coinvolga un target nel tessuto vivente, tanto meno che l’interazione spieghi analgesia, ansia o infiammazione.
Differenze di specie, metaboliti ed effetti di membrana
Anche segnali forti in vitro possono fallire fuori dalla piastra perché la farmacologia dei cannabinoid è altamente dipendente dal contesto. I recettori umani e murini non sono sempre funzionalmente identici. Un composto attivo su TRPA1 murino o su readout legati a 5-HT1A nel ratto può cambiare potenza o efficacia nell’ortologo umano. Lo stesso avvertimento vale per le varianti di splicing, la riserva recettoriale e il background cellulare. Una linea con recettori fortemente sovraespressi può far sembrare importante un ligando debole.
Il metabolismo aggiunge un ulteriore livello. Molti cannabinoid non restano a lungo nella forma madre. THC diventa 11-hydroxy-THC; altre strutture formano metaboliti ossidati o coniugati che possono differire nettamente nel profilo di target. I regolatori stanno prestando maggiore attenzione a questo problema nei dibattiti affini sulla politica del farmaco. Nel 2025, HHS ha affermato che “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety,” evidenziando una lezione più ampia: gli intossicanti potenziati o metabolicamente avvantaggiati possono comportarsi in modo molto diverso dal composto madre. Il campo del cannabis ha problemi paralleli con cannabinoid semi-sintetici, nuovi isomeri e formulazioni progettate per spostare l’esposizione. Se si saggia solo la molecola madre, si può perdere la specie che davvero guida gli effetti in vivo.
Le membrane sono la confusione silenziosa più grande. I cannabinoid sono abbastanza lipofili da accumularsi nei doppi strati e nei compartimenti intracellulari. Ciò significa che la concentrazione nominale nel bagno è spesso un pessimo proxy della concentrazione nel target. Una applicazione a 10 micromolari in un saggio cellulare può creare un carico di membrana localmente molto alto, alterando in modo aspecifico il gating del canale o il comportamento del recettore. Può anche produrre falsi negativi se la concentrazione acquosa libera è molto più bassa del previsto perché il composto si attacca alla plastica, alle proteine sieriche o alla membrana stessa.
Ecco perché le affermazioni ad alte concentrazioni meritano scetticismo. Se un cannabinoid influenza un target solo oltre i 20 o 30 micromolari, la prima domanda dovrebbe essere se questo rifletta un’interazione fisiologicamente rilevante o un artefatto indotto dalla membrana. I canali TRP sono particolarmente vulnerabili all’esagerazione qui. Sono target genuinamente responsivi ai cannabinoid in alcuni casi, ma gli effetti possono essere bifasici, rapidamente desensibilizzanti e altamente sensibili alla concentrazione. Un breve picco di calcio ad alte concentrazioni micromolari non significa automaticamente un meccanismo terapeuticamente rilevante.
Dalle pubblicazioni ACS sul docking alla farmacologia reale
La chimica medicinale prospera nella semplificazione, ma la biologia punisce la semplificazione eccessiva. L’articolo sul docking per CB2 mostra la versione utile della semplificazione: partire da una struttura recettoriale, fare screening di librerie, prioritizzare candidati, sintetizzare analoghi, testarli in saggi di legame e funzionali e poi imparare dalle relazioni struttura-attività. Questa sequenza può generare veri farmaci. Può anche rivelare che la posa in silico più attraente appartiene a un composto con scarsa permeabilità, metabolismo instabile, segnalazione biasata o nessuna attività significativa nelle cellule native.
La distanza dalla posa docked alla farmacologia è esattamente il punto in cui molte affermazioni sui cannabinoid falliscono. Un’immagine di docking di CBD o CBG in TRPV1, 5-HT1A, GPR55 o PPAR-gamma non è la prova che il composto guidi la fisiologia rilevante in animali o esseri umani. La farmacologia reale richiede convergenza. Idealmente significa impegno diretto del target a concentrazioni plausibili, effetti funzionali in sistemi nativi, evidenze di loss-of-function usando antagonisti o knockout, supporto farmacocinetico e un collegamento al comportamento o all’esito clinico.
Quando questi livelli si allineano, la storia diventa rapidamente più forte. L’attuale spinta di ricerca a separare analgesia e intossicazione dipende da questa logica. Il report del 2026 di ScienceDaily che descrive “a cannabis compound that relieves pain without the high” è interessante proprio perché va oltre l’equivalenza grossolana con THC e punta verso meccanismi selettivi per target o perifericamente ristretti. Lo stesso vale per il lavoro sui canali del sodio di THC. Lo stesso vale per gli sforzi di costruire ligandi selettivi tramite docking e progettazione guidata dalla struttura. Il pattern è coerente anche quando singole affermazioni richiedono correzioni.
I lettori dovrebbero esigere la stessa disciplina dagli articoli accademici sui cannabinoid. Chiedere quale saggio sia stato usato. Chiedere se la concentrazione attiva sia realistica. Chiedere se l’effetto sopravvive all’elettrofisiologia, agli antagonisti, ai knockout, agli studi di metabolismo e alla traslazione di specie. Soprattutto, chiedere se la tesi sul target spieghi l’organismo, non solo la piastra. È così che la farmacologia cannabinoid non CB1/CB2 smette di essere una raccolta di indizi interessanti e diventa un vero meccanismo.
Livelli di evidenza: dal piatto cellulare alla clinica
La letteratura sui target non CB1/CB2 è ricca perché i cannabinoid sono chimicamente promiscui. Una singola molecola può colpire TRPV1, segnalazione legata a 5-HT1A, PPAR-gamma, GPR55, tono dell’adenosina e canali del sodio voltaggio-dipendenti a seconda di concentrazione, tessuto e profilo metabolico. Questo produce articoli meccanicistici interessanti. Non produce automaticamente medicina provata. Se esiste una regola che mantiene questo campo onesto, è semplice: ogni passo in più sulla scala delle evidenze elimina una grande quota di affermazioni che sembravano convincenti a livello inferiore.
Cosa può e non può provare l’evidenza preclinica
Alla base della scala ci sono i saggi di legame, le registrazioni dei canali, i sistemi reporter e le colture cellulari. Questi metodi sono indispensabili. È grazie a essi che i ricercatori hanno imparato che la farmacologia dei cannabinoid si estende ben oltre i recettori classici, ed è per questo che le spiegazioni riduzioniste sul cannabis appaiono ora datate. Il premio Nobel 2021 ha riconosciuto David Julius e Ardem Patapoutian “for their discoveries of receptors for temperature and touch”, ricordando che la biologia TRP non è trivia periferica; si colloca vicino al centro della moderna scienza del dolore. Quando i cannabinoid attivano o desensibilizzano TRPV1, TRPA1 o canali correlati in una piastra, quel risultato conta.
Ma una piastra non può dirti se il medesimo coinvolgimento del target avvenga a dosi umane tollerate. Molti effetti in vitro dei cannabinoid appaiono solo a concentrazioni micromolari. I livelli plasmatici dopo dosaggio nel mondo reale possono essere più bassi, più transitori o spostati verso metaboliti con farmacologia diversa. CBD è l’esempio classico. Mostra ripetutamente azioni difficili da spiegare solo tramite CB1 o CB2, motivo per cui TRPV1, 5-HT1A, PPAR-gamma, GPR55 e le vie dell’adenosina continuano ad apparire in letteratura. Eppure il fatto che CBD possa modulare un target in cellule coltivate non dimostra che questo meccanismo guidi un esito clinico in epilessia, ansia, dolore o infiammazione.
I modelli animali sono un passo più vicino alla medicina e ancora lontani da essa. Gli studi sui roditori possono mostrare che un cannabinoid riduce l’allodinia, sopprime i marcatori infiammatori o modifica il comportamento simile all’ansia. Possono persino supportare racconti specifici di target con antagonisti, knockout o restrizione periferica. Il recente report della Hebrew University è un buon esempio di perché questo lavoro sia entusiasmante: i ricercatori hanno riportato che THC inibisce i nocicettori periferici prendendo di mira i canali del sodio nocicettivi NaV1.7 e NaV1.8. Questo risultato va contro l’assunzione pigra secondo cui l’analgesia da THC debba essere solo una storia di CB1 più intossicazione. Se l’effetto reggerà, suggerisce un’azione rilevante per il dolore al livello stesso dell’eccitabilità periferica.
Resta comunque vero che anche un forte lavoro animale non può provare che bloccare NaV1.7/NaV1.8 con un cannabinoid diventerà un analgesico umano sicuro ed efficace. Le differenze di specie contano. Il dosaggio conta. La via conta. I readout comportamentali nei topi possono ingannare. Un segnale di dolore attenuato in una preparazione nervosa o in un test alla formalina può non tradursi in sollievo per neuropatia, artrosi o dolore post-operatorio nell’uomo. La stessa cautela vale per gli annunci aziendali. Nel 2025, MIRA Pharmaceuticals ha affermato che il candidato MIRA-55 mostrava un “differentiated mechanism of action” e “anxiolytic activity relative to THC” in dati preclinici. Questo è legittimo come evidenza di ricerca. Non è una rivendicazione terapeutica, e non dovrebbe essere trattata come tale.
La stessa cautela traslazionale vale per i titoli attraenti. ScienceDaily nel 2026 ha evidenziato lavori su “a cannabis compound that relieves pain without the high.” Forse. È una direzione utile per la progettazione del farmaco, specialmente se target periferici o meccanismi non CB1 possono separare analgesia e intossicazione centrale. Ma “without the high” in un report preclinico è un’ipotesi in prova, non un fatto consolidato sulla terapia umana.
Medicina cannabinoid approvata e ristrettezza delle indicazioni attuali
Ora salire alla cima della scala: evidenza da farmaco approvato. Qui il campo diventa molto più stretto. L’esempio statunitense più chiaro è la soluzione orale di cannabidiol. L’etichetta approvata dalla FDA afferma che è indicata “for the treatment of seizures associated with Lennox-Gastaut syndrome, Dravet syndrome, or tuberous sclerosis complex in patients 1 year of age and older.” Questa frase è più informativa di decine di vaghe affermazioni di benessere perché nomina la forma farmaceutica, l’esito, le malattie e la fascia d’età.
Si noti cosa non dice l’etichetta. Non dice che CBD sia approvato per dolore, ansia generalizzata, insonnia, malattia infiammatoria intestinale, neuroprotezione o un generico “equilibrio endocannabinoid.” Non convalida ogni meccanismo proposto nella letteratura preclinica. L’approvazione ci dice che un prodotto specifico di cannabidiol ha dimostrato efficacia e sicurezza accettabile per disturbi convulsivi specifici in studi controllati. Non risolve se TRPV1, GPR55, 5-HT1A, PPAR-gamma, gli effetti sul calcio intracellulare o le azioni dei metaboliti siano il meccanismo clinico dominante. Il meccanismo può restare in parte irrisolto anche quando l’efficacia è reale.
Questo scarto tra indicazione approvata e folklore meccanicistico è comune in farmacologia. Molti farmaci hanno funzionato nei pazienti prima che la loro mappa completa dei target fosse compresa. I cannabinoid non sono insoliti sotto questo profilo. Ciò che è insolito è quanto spesso le affermazioni ampie vengano ricostruite a posteriori da risultati recettoriali sparsi e poi presentate come se avessero già superato i test clinici.
La ristrettezza delle indicazioni attuali conta anche nelle discussioni di salute pubblica. I regolatori distinguono sempre più i cannabinoid vegetali familiari dagli intossicanti alterati, potenziati o semi-sintetici con profili di rischio diversi. Nel 2025, HHS ha affermato che “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” sostenendo un’azione di scheduling sui prodotti 7-OH potenziati. La dichiarazione riguardava prodotti correlati al kratom, non il cannabis, ma la lezione politica si trasferisce bene: quando i produttori iniziano ad arricchire, modificare o sintetizzare molecole psicoattive potenti, le scorciatoie basate sull’origine vegetale diventano inaffidabili. La farmacologia a livello di target comincia a contare molto.
Perché le storie meccanicistiche spesso superano i dati clinici
Li superano perché le storie meccanicistiche sono rapide, vivide e pubblicabili. La prova clinica è lenta e costosa. Un articolo cellulare può mostrare che un cannabinoid attiva TRPA1, antagonizza GPR55 o modifica la segnalazione di 5-HT1A in un progetto di mesi. Un trial randomizzato convincente per il dolore cronico può richiedere anni e fallire comunque perché l’effetto è piccolo, gli effetti avversi limitano il dosaggio o il target preclinico non è mai stato realmente coinvolto nei pazienti.
La chimica dei cannabinoid invita anche alla sovrainterpretazione. Composti strutturalmente correlati possono avere profili di target molto diversi, e il metabolismo può riscrivere il quadro dopo la somministrazione. La via cambia di nuovo la storia. Il dosaggio orale produce metaboliti di primo passaggio; l’inalazione cambia la cinetica; formulazioni topiche o periferiche possono favorire target locali anziché centrali. Persino la categoria legale “hemp” dice poco dal punto di vista farmacologico. Il Farm Bill del 2018 ha tracciato la linea a delta-9 THC “not more than 0.3 percent on a dry weight basis,” una soglia statutaria, non biologica.
La lettura corretta delle evidenze non CB1/CB2 è quindi né rifiuto né hype. La letteratura preclinica mostra davvero che i cannabinoid agiscono su più di CB1 e CB2. Per il dolore in particolare, i canali TRP e i canali del sodio meritano seria attenzione. Per CBD, i target non classici non sono note accessorie; sono probabilmente centrali per spiegare il suo profilo. Ma plausibilità non equivale a efficacia, l’impegno del target non equivale a beneficio per il paziente, e l’approvazione di un solo farmaco cannabinoid per un piccolo gruppo di disturbi convulsivi non ratifica la nube molto più ampia di affermazioni costruite su diagrammi recettoriali, comportamento murino e risultati su piastra cellulare.
Sicurezza, regolamentazione e perché la farmacologia off-target conta per la salute pubblica
I problemi di salute pubblica non si allineano ordinatamente con i diagrammi dei recettori. Un cannabinoid può essere di origine vegetale, derivato da hemp, semi-sintetico o pienamente sintetico e tuttavia produrre rischi che sono scarsamente prevedibili dalla sua etichetta, dalla sua origine o dalla sua categoria legale. Questo è il significato pratico della farmacologia off-target. Una volta che una molecola tocca canali TRP, recettori della serotonina, PPAR, sistemi di segnalazione simili a GPR55, canali del sodio e altri target non CB1/CB2, il profilo di sicurezza può cambiare in modi rilevanti per la sorveglianza delle intossicazioni, gli standard di prodotto, le politiche sulla guida alterata e il rischio di dipendenza.
L’errore non è solo scientifico. È regolatorio. La politica sul cannabis ha spesso trattato “cannabinoid” come se fosse già una spiegazione.
Intossicanti potenziati e la lezione politica da 7-OH
L’avvertimento più chiaro oggi non proviene nemmeno da un costituente classico del cannabis. Nel 2025, il Department of Health and Human Services degli Stati Uniti ha affermato che “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” sostenendo l’azione della DEA sui prodotti 7-OH potenziati. Questa frase conta perché mostra le autorità sanitarie federali tracciare una linea tra l’esposizione botanica familiare e gli intossicanti concentrati o chimicamente manipolati che possono comportarsi in modo molto diverso nelle popolazioni reali.
La lezione politica si trasferisce direttamente ai cannabinoid. Una categoria ampia come “hemp-derived”, “plant-based” o persino “cannabinoid-like” non dice ai regolatori quasi nulla sui target che un composto coinvolge alle concentrazioni rilevanti per l’uso. Non dice ai consumatori quasi nulla nemmeno su potenza, inizio dell’effetto, durata, rischio di interazione o potenziale di abuso.
I prodotti 7-OH potenziati sono diventati un problema di salute pubblica perché la chimica ha cambiato l’esposizione. Quando un mercato passa da una presenza naturale in tracce a un ingrediente attivo concentrato, la farmacologia smette di essere una curiosità e diventa la questione centrale di sicurezza. Lo stesso schema si è già visto attorno ai cannabinoid semi-sintetici e strutturalmente modificati venduti sotto l’ombrello della legalità hemp dopo che il Farm Bill del 2018 ha definito hemp con una concentrazione di delta-9 THC di “not more than 0.3 percent on a dry weight basis.” Questa soglia sul peso secco è una definizione agricola, non uno standard di farmacologia. Non seleziona attività TRP, blocco dei canali del sodio, segnalazione di 5-HT1A, effetti di GPR55 o metaboliti con persistenza più lunga e diversa penetrazione nel SNC.
Ecco perché gli effetti off-target non sono una nota oscura. Sono una via attraverso cui prodotti commercializzati come vicini al cannabis possono generare danni inattesi. Un composto presentato pubblicamente come “THC-like but legal” può differire nell’efficacia intrinseca su CB1, ma anche in modi molto meno visibili: maggiore stimolazione cardiovascolare, più forte potenziale pro-convulsivante o ansiogeno, disforia più severa, sedazione insolita o un profilo tossicologico modellato dal metabolismo e non solo dal farmaco madre. Queste possibilità non sono ipotetiche in astratto; sono esattamente il motivo per cui le agenzie sanitarie reagiscono in modo diverso agli intossicanti potenziati rispetto alle categorie botaniche ampie.
Una risposta regolatoria sensata parte da domande a livello di target. Quali recettori e canali sono coinvolti? A quali concentrazioni? In quali tessuti? Quali sono i metaboliti principali? Ci sono evidenze di inibizione dei canali del sodio che possano modificare nocicezione o conduzione cardiaca? L’attivazione di TRPV1 è probabilmente in grado di desensibilizzare la segnalazione del dolore o di irritare e peggiorare i sintomi a esposizioni più basse o transitorie? Sono domande più difficili di “è un cannabinoid”, ma sono quelle giuste.
I limiti del pensiero basato sull’equivalenza con THC
L’equivalenza con THC è attraente perché semplifica diritto, tassazione, etichettatura e discussioni sull’alterazione cognitiva. È anche spesso errata. Due composti possono produrre un certo grado di intossicazione e comunque differire nettamente per rischio ansiogeno, potenziale psicotomimetico, effetto analgesico, risposta sulla frequenza cardiaca, controllo dell’emesi, sviluppo della tolleranza e carico di astinenza perché non condividono la stessa mappa di target più ampia.
Anche THC stesso non è farmacologicamente esaurito da CB1 e CB2. Nel 2025, i ricercatori della Hebrew University hanno riportato che THC inibisce i nocicettori periferici prendendo di mira i canali del sodio nocicettivi NaV1.7 e NaV1.8. Questo risultato va contro la narrazione semplicistica “THC agisce qui, CBD agisce lì.” Se persino un cannabinoid psicoattivo canonico può influire direttamente sui canali del sodio voltaggio-dipendenti rilevanti per il dolore, allora sicurezza ed efficacia non possono essere inferite solo dal marchio cannabinoid. Dose, via, distribuzione ed esposizione tissutale diventano decisive.
Lo stesso punto emerge in senso opposto con CBD. La sua soluzione orale approvata è indicata dalla FDA per convulsioni associate alla sindrome di Lennox-Gastaut, alla sindrome di Dravet o al complesso della sclerosi tuberosa in pazienti di 1 anno e oltre, un fatto clinico che non è mai stato ben spiegato dall’agonismo di CB1 perché CBD non è un intossicante CB1 diretto. Il suo profilo ha da tempo spinto i ricercatori verso altri meccanismi, tra cui TRPV1, 5-HT1A, segnalazione correlata all’adenosina e PPAR-gamma. Non tutti questi meccanismi sono ugualmente consolidati nell’uomo, ma insieme rendono inevitabile un punto: gli effetti dei cannabinoid spesso si collocano in una rete, non in un singolo interruttore recettoriale.
Questa visione di rete spiega anche perché l’intossicazione sia una metrica pessima per la salute pubblica. Un prodotto può essere meno intossicante di THC ma comunque produrre interazioni farmacologiche preoccupanti, effetti sugli enzimi epatici, stress cardiovascolare, reazioni di panico o sedazione. Può anche essere più intossicante senza essere più prevedibile. Il pubblico spesso sente “più debole di THC” o “più forte di THC” come se questo risolvesse il problema. Non è così. Queste sono di solito affermazioni su un fenotipo evidente, non su un profilo tossicologico completo.
La pipeline di ricerca si sta già muovendo oltre l’equivalenza con THC. Un comunicato Nasdaq del 2025 di MIRA Pharmaceuticals descriveva dati preclinici per MIRA-55 con un “differentiated mechanism of action” e attività ansiolitica rispetto a THC. I comunicati aziendali sono prove deboli rispetto ai dati clinici peer-reviewed, quindi l’affermazione va trattata con cautela. Tuttavia, la direzione è reale: la chimica medicinale sta cercando di separare gli effetti desiderati dall’intossicazione centrale cambiando l’impegno del target. La stessa logica traslazionale appare in un report del 2026 di ScienceDaily che descrive un lavoro su “a cannabis compound that relieves pain without the high.” I risultati allo stadio di ricerca non sono prova clinica, ma rafforzano un punto regolatorio centrale. Se gli effetti benefici possono essere separati dall’intossicazione, allora anche i danni possono esserlo. Un prodotto a basso high non è automaticamente un prodotto a basso rischio.
Perché i nuovi cannabinoid richiedono esame a livello di target
I nuovi cannabinoid meritano più scrutinio, non meno, proprio perché vengono spesso introdotti sul mercato prima che la loro farmacologia nell’uomo sia mappata. La scorciatoia vecchia era chiedere se una molecola si legasse a CB1 o CB2. La domanda migliore è cos’altro faccia, e se tali azioni diventino rilevanti a dosi reali dopo inalazione, ingestione orale o metabolismo.
Per il dolore, i canali TRP e i canali del sodio sono esempi ovvi. David Julius e Ardem Patapoutian hanno ricevuto il premio Nobel per la Fisiologia o Medicina 2021 per scoperte dei recettori per temperatura e tatto, un promemoria che la biologia somatosensoriale è costruita su target che i cannabinoid possono influenzare al di fuori dei recettori cannabinoid classici. L’attivazione di TRPV1 può contribuire all’analgesia attraverso la desensibilizzazione, ma può anche produrre irritazione ed è fortemente dipendente dalla concentrazione. Un regolatore che guardi solo all’attività CB1 perderà interamente quell’asse rischio-beneficio.
Per la sicurezza psichiatrica, la segnalazione della serotonina conta. CBD è stato ripetutamente discusso come candidato ansiolitico legato a 5-HT1A, ma il coinvolgimento serotoninergico può essere indiretto e dipendente dal contesto. Questa incertezza non è un motivo per ignorare il target; è un motivo per studiarlo attentamente prima che i prodotti vengano normalizzati come innocui. Lo stesso vale per GPR55, talvolta proposto come candidato “CB3” nonostante il dibattito in corso. Un target contestato resta comunque un target che può modellare il calcio, l’eccitabilità e le risposte infiammatorie.
Per gli effetti metabolici e infiammatori, target intracellulari come PPAR-gamma complicano ulteriormente il quadro. La segnalazione dei recettori nucleari non assomiglia all’effetto intossicante rapido, eppure può essere rilevante per esposizione cronica, distribuzione nel tessuto adiposo, cambiamenti trascrizionali e interazioni con altri stati patologici. La comunicazione di salute pubblica costruita intorno a se qualcosa “gives you high” perde questi rischi più lenti e silenziosi.
Qui regolazione, tossicologia e comunicazione della sicurezza per i consumatori hanno bisogno di alfabetizzazione sui target molecolari. Non tutti i colpi in vitro conteranno clinicamente. Le differenze di specie sono reali. I metaboliti possono dominare. Ma le agenzie dovrebbero richiedere pannelli di target, caratterizzazione dei metaboliti, dati concentrazione-risposta e studi umani rilevanti per l’alterazione cognitiva prima di assumere che un nuovo cannabinoid possa essere regolato come un altro. L’articolo del 2016 dell’ACS Journal of Medicinal Chemistry “Library Docking for Cannabinoid-2 Receptor Ligands” riflette il modo in cui oggi funziona il drug discovery: progettazione basata sulla struttura, ingegneria della selettività e attenzione esplicita alle differenze a livello recettoriale. La regolamentazione dovrebbe smettere di fingere che il mercato sia più semplice di quanto la chimica medicinale sappia già che sia.
La salute pubblica non può permettersi il riduzionismo recettoriale. “Cannabinoid” è un’etichetta di partenza. Non è una conclusione di sicurezza.
Drug discovery: progettare cannabinoid e molecole ispirate ai cannabinoid per target non CB1/CB2
Il drug discovery attorno ai cannabinoid si è spinto ben oltre la vecchia domanda se una molecola sia “attiva su CB1” o “attiva su CB2.” Questa semplificazione era sempre fragile, perché molti composti correlati ai cannabinoid sono farmacologicamente confusi: colpiscono canali ionici, GPCR oltre la coppia canonica, recettori nucleari intracellulari, processi di trasporto ed enzimi metabolizzanti, spesso con effetti diversi a seconda di concentrazione, tessuto e via di somministrazione. Per i chimici medicinali, quel disordine non è solo un problema. È anche un’opportunità.
L’obiettivo centrale del design è chiaro abbastanza: mantenere gli effetti analgesici, anti-infiammatori o ansiolitici riducendo le responsabilità legate alla forte attivazione di CB1 nel cervello. Queste responsabilità non sono astratte. Sedazione, compromissione cognitiva, intossicazione, potenziale di abuso ed effetti psichiatrici limitanti la dose sono esattamente il motivo per cui “THC-like” è spesso un profilo cattivo in un programma di sviluppo, anche quando THC stesso mostra una farmacologia utile. L’attuale clima regolatorio rafforza questo punto. Nel 2025, HHS ha sostenuto un’azione di scheduling affermando che “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety”, un promemoria che gli intossicanti modificati o potenziati non possono essere considerati come i composti vegetali familiari solo perché sono vicini su uno scaffale di marketing. La farmacologia a livello di target conta.
Restrizione periferica, selettività funzionale e segnalazione biasata
Una via per aggirare gli effetti avversi centrali è brutale ma efficace: tenere il farmaco fuori dal cervello. La restrizione periferica può essere progettata aumentando la superficie polare, incrementando la capacità di formare legami a idrogeno, regolando il pKa o rendendo la molecola un substrato per i trasportatori di efflusso alla barriera ematoencefalica. L’idea non è unica della scienza cannabinoid, ma si adatta in modo particolarmente buono al campo perché la segnalazione del dolore spesso inizia nei nocicettori periferici, nei tessuti infiammati e nei gangli della radice dorsale.
È qui che i target non CB1/CB2 diventano particolarmente attrattivi. Un report del 2025 della Hebrew University ha sostenuto che THC inibisce i nocicettori periferici prendendo di mira i canali del sodio NaV1.7 e NaV1.8, due grandi motori dell’eccitabilità nocicettiva. Se questo meccanismo reggerà in più sistemi, la sua importanza è enorme. NaV1.7 è da tempo considerato un target di primo piano per il dolore perché le mutazioni loss-of-function umane in SCN9A possono produrre una profonda insensibilità congenita al dolore. Uno scheletro cannabinoid che preservi la modulazione dei canali del sodio in periferia minimizzando al contempo la segnalazione centrale di CB1 non sarebbe semplicemente un “THC meno intossicante.” Sarebbe un analgesico di tipo diverso.
I canali TRP creano una finestra analoga. La biologia sensoriale più ampia qui è stata riconosciuta al massimo livello quando il premio Nobel per la Fisiologia o Medicina 2021 è andato a David Julius e Ardem Patapoutian per scoperte dei recettori per temperatura e tatto. TRPV1, TRPA1 e canali correlati sono profondamente legati a nocicezione e segnalazione infiammatoria, e diversi fitocannabinoid interagiscono con essi in modi sensibili alla concentrazione e talvolta paradossali: l’attivazione iniziale può essere seguita da desensibilizzazione, con quest’ultima che potenzialmente contribuisce all’analgesia. Ciò rende la chimica medicinale più difficile, non più facile. Ma significa anche che un composto non deve essere un ligando ortosterico pulito on-off dei recettori CB per avere valore terapeutico.
La selettività funzionale aggiunge un ulteriore livello. Anche su CB1 o CB2 stessi, i ligandi possono favorire un output di segnalazione rispetto a un altro, spostando l’equilibrio tra vie della proteina G, reclutamento di beta-arrestina, internalizzazione del recettore e programmi trascrizionali a valle. In termini semplici, due molecole possono entrambe “legarsi a CB1” e tuttavia comportarsi in modo molto diverso nel tessuto vivente. L’articolo del 2016 del Journal of Medicinal Chemistry “Library Docking for Cannabinoid-2 Receptor Ligands” riflette quanto la strategia di design si sia spostata verso la regolazione basata sulla struttura invece che sulle etichette recettoriali grossolane. La stessa logica ora si estende oltre i recettori CB: i chimici vogliono scaffold la cui forma, lipofilia e vincoli conformazionali li orientino verso un meccanismo rilevante per il dolore o l’ansia evitando il pesante bagaglio della segnalazione centrale.
CBD è la prova stabile che un farmaco correlato ai cannabinoid può avere rilevanza clinica senza essere spiegato dall’agonismo di CB1/CB2. L’etichettatura FDA per la soluzione orale di cannabidiol, aggiornata nel 2024, copre le convulsioni associate alla sindrome di Lennox-Gastaut, alla sindrome di Dravet e al complesso della sclerosi tuberosa in pazienti di 1 anno e oltre. Qualunque combinazione di TRPV1, 5-HT1A, GPR55-related, vie adenosinergiche, effetti intracellulari e di rete spieghi infine quell’efficacia, non è una semplice storia CB1. I ricercatori l’hanno notato.
MIRA-55 e la spinta verso meccanismi differenziati
MIRA-55 è un utile caso di studio, non perché chiuda la questione, ma perché mostra come le aziende ora inquadrino i programmi cannabinoid. In un comunicato diffuso da Nasdaq nel 2025, MIRA Pharmaceuticals ha affermato che il proprio candidato mostrava un “differentiated mechanism of action” e “anxiolytic activity relative to THC” nel lavoro preclinico. Questa formulazione dice quasi tutto sul segnale attuale per investitori e regolatori. Essere ispirati ai cannabinoid non basta più. Un’azienda vuole distanza dalla semplice imitazione di THC, specialmente per indicazioni come l’ansia, dove gli effetti avversi centrali possono cancellare il beneficio.
Resta comunque indispensabile lo scetticismo. “Differentiated mechanism” in un comunicato aziendale è una tesi, non una conclusione. Può significare un coinvolgimento recettoriale alterato, distribuzione tissutale cambiata, metaboliti distinti, agonismo parziale, bias funzionale, effetti off-target sui canali ionici o semplicemente un profilo comportamentale diverso in un test animale. Senza pannelli farmacologici completi, curve concentrazione-risposta, identificazione dei metaboliti, dati di occupazione recettoriale e replicazione in cieco, la frase è più un’ipotesi che un risultato.
Detto ciò, la strategia dietro l’affermazione è credibile. Se un composto può ridurre il comportamento simile all’ansia abbassando intossicazione, compromissione della memoria o soppressione locomotoria rispetto a THC, la chimica medicinale probabilmente ha cambiato una o più di quattro cose: penetrazione cerebrale, efficacia intrinseca su CB1, coinvolgimento di target non CB come 5-HT1A o canali TRP, o conversione metabolica in specie attive con un profilo di target diverso. Sono esattamente gli assi su cui competono i programmi cannabinoid moderni.
MIRA-55 illustra anche il problema della deconvoluzione dei target. Le molecole simili ai cannabinoid sono spesso “sporche” in senso farmacologico. Questo non è un giudizio morale; è un avvertimento meccanicistico. Se emerge un segnale ansiolitico preclinico, non si può assumere che un singolo recettore lo spieghi. La segnalazione serotoninergica può essere diretta o indiretta. Gli effetti PPAR-gamma possono richiedere accumulo intracellulare o metaboliti. GPR55 può sembrare importante in un saggio e marginale in un altro. Un colpo in vitro a 10 micromolari può essere irrilevante se le concentrazioni cerebrali libere non si avvicinano mai a quel livello in vivo.
Come appare una pipeline realistica per i cannabinoid
Una pipeline realistica è più stretta e disciplinata di quanto suggerisca la retorica pubblica. Non è una sfilata di “non-psychoactive cannabis compounds” diretti all’approvazione. È un filtro.
All’inizio ci sono modificazione dello scaffold e triage: cannabinoid classici, cannabinoid non classici, lipidi ispirati agli endocannabinoid e chemotipi non correlati che imitano una sola caratteristica utile della farmacologia cannabinoid senza ereditare l’intero pacchetto. I chimici alterano la lunghezza della catena laterale, i vincoli dell’anello, la stereochimica, la posizione degli eteroatomi e i punti fragili del metabolismo, quindi testano non solo CB1 e CB2 ma anche TRPV1, TRPA1, canali NaV, alcuni GPCR orfani e saggi rilevanti per la serotonina. I candidati che emergono vengono poi sottoposti a studi ADME, analisi della concentrazione non legata e misurazioni cervello-plasma. Molti falliscono lì.
I programmi che sopravvivono si dividono di solito in alcune categorie credibili. Una sono gli analgesici a restrizione periferica, dove il sogno è il sollievo dal dolore tramite canali del sodio, desensibilizzazione TRP, modulazione infiammatoria o segnalazione cannabinoid periferica senza esposizione sostanziale al SNC. Un’altra sono i ligandi CB1 biasati o a bassa efficacia, che mirano a preservare la segnalazione terapeutica riducendo l’intossicazione. Una terza sono i composti a meccanismo misto, spesso più realistici del purismo a target singolo, dove azioni modeste su più nodi del dolore o dell’ansia superano un ligando “pulito” ma clinicamente debole.
Il gancio traslazionale che mantiene viva quest’area è semplice: la separazione dell’analgesia dal high sembra possibile, almeno nei sistemi di ricerca. Il riassunto 2026 di ScienceDaily su nuovi lavori che descrivono “a cannabis compound that relieves pain without the high” va trattato con attenzione, perché i titoli superano i dati, ma il concetto si inserisce nella direzione più ampia della chimica medicinale. Lo stesso vale per il lavoro sui canali del sodio di THC. Lo stesso vale per gli sforzi di costruire ligandi selettivi tramite docking e progettazione guidata dalla struttura. Il pattern è coerente anche quando le singole affermazioni necessitano di essere limate.
Quello che probabilmente la pipeline non sarà, almeno a breve, è un successo clinico di massa. Promiscuità di target, complessità dei metaboliti, differenze di specie ed effetti di formulazione continuano a rompere le storie semplici. Ma il campo non è più bloccato a chiedersi se i cannabinoid siano “davvero” una questione di CB1 e CB2. I drug developer hanno già risposto a questa domanda con i loro budget di chimica. Stanno progettando per lo spazio oltre quei recettori, perché è lì che si trova ormai la migliore possibilità di separare beneficio e responsabilità.
Convinzioni errate comuni e controversie irrisolte
L’errore più grande nella discussione pubblica sui cannabinoid è il riduzionismo recettoriale: la spinta a forzare una farmacologia confusa in un target principale da titolo. Questa abitudine era sempre fragile, e sta diventando più rischiosa mentre regolatori e sviluppatori di farmaci affrontano composti che non sono più soltanto “THC” o “CBD” nel senso semplice, derivato dalla pianta. Nel 2025, HHS ha affermato che “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” sostenendo un’azione della DEA sui prodotti 7-OH potenziati. Classe di farmaco diversa, stessa lezione: una volta che i chimici alterano, arricchiscono o semi-sintetizzano uno scheletro intossicante, le vecchie assunzioni sull’azione recettoriale e sulla sicurezza possono crollare rapidamente. La scienza dei cannabinoid ha lo stesso problema. Un cannabinoid non è una chiave magica per una sola serratura. È di solito un ligando promiscuo i cui effetti nel mondo reale dipendono da concentrazione, esposizione tissutale, metabolismo e dai target secondari che diventano rilevanti a quei livelli.
Esiste davvero un recettore CB3
Non esiste alcun recettore CB3 condiviso dal consenso.
Questa risposta suona netta perché le evidenze la richiedono. Nel corso degli anni sono stati proposti diversi recettori come candidati “CB3”, soprattutto GPR55, talvolta GPR18 e occasionalmente altri GPCR orfani che rispondono in alcuni sistemi di saggio a ligandi cannabinoid o a lipidi correlati agli endocannabinoid. Ma un target proposto non è una classe recettoriale accettata. CB1 e CB2 hanno ottenuto il loro nome attraverso evidenze convergenti: clonazione, farmacologia riproducibile dei ligandi, distribuzione tissutale, segnalazione e ampia replicazione. I candidati a CB3 non hanno mai superato quella soglia.
GPR55 è il sospettato abituale. È espresso nel cervello, nelle cellule immunitarie, nell’intestino e nell’osso, e alcuni cannabinoid vi interagiscono. CBD è stato spesso descritto come antagonista di GPR55 nei saggi cellulari; alcuni cannabinoid sintetici mostrano attività anch’essi. Tuttavia, la farmacologia è incoerente tra laboratori, ligandi e readout. Alcuni composti appaiono attivi in un saggio di segnalazione e silenziosi in un altro. Le differenze di specie complicano il quadro. I ligandi endogeni sono dibattuti. Soprattutto, chiamare GPR55 “CB3” suggerisce un posto definito all’interno della famiglia canonica dei recettori cannabinoid che il campo non gli ha ancora assegnato.
Questo importa perché le etichette possono correre più veloci delle evidenze. Una volta che un recettore riceve un soprannome accattivante, il soprannome inizia a svolgere un lavoro esplicativo che non ha ancora guadagnato. Dolore? CB3. Ansia? CB3. Effetti sull’osso? CB3. Questa non è farmacologia; è branding. La posizione più prudente è che GPR55, GPR18, GPR119 e recettori correlati siano target non CB1/CB2 con diversi livelli di evidenza per la modulazione da parte di cannabinoid o lipidi simili ai cannabinoid. Alcuni possono contare molto in tessuti specifici. Nessuno ha ottenuto uno status consensuale come “il terzo recettore cannabinoid.”
Il drug discovery si è mosso di conseguenza. Gli articoli di chimica medicinale non si comportano come se un nuovo nome di recettore risolvesse il sistema. L’articolo del 2016 sul Journal of Medicinal Chemistry “Library Docking for Cannabinoid-2 Receptor Ligands” è un buon indicatore di dove il campo si trova davvero: progettazione basata sulla struttura, selettività recettoriale, ingegneria dello scaffold e ottimizzazione diretta dal target, non mitologia su un misterioso CB3 in attesa di spiegare tutto. Allo stesso modo, le affermazioni delle aziende sui composti cannabinoid di nuova generazione ora spesso enfatizzano meccanismi differenziati piuttosto che una semplice agonismo recettoriale più forte. Il comunicato preclinico 2025 di MIRA Pharmaceuticals per MIRA-55 affermava esplicitamente un “differentiated mechanism of action” e attività ansiolitica rispetto a THC. Si tratta di materiale promozionale, non scienza consolidata, ma riflette un vero spostamento strategico: i cannabinoid terapeutici utili potrebbero derivare dall’allontanarsi dalla semplice intossicazione CB1, non dalla scoperta di un recettore jolly.
CBD agisce principalmente attraverso la serotonina
Anche no, anche se la segnalazione serotoninergica fa parte della storia.
CBD viene spesso commercializzato nella cultura pop come se fosse sostanzialmente un farmaco naturale 5-HT1A. Questa semplificazione sopravvive perché dietro c’è evidenza reale. In studi preclinici, CBD ha mostrato effetti ansiolitici e anti-stress che in alcuni paradigmi vengono ridotti dagli antagonisti di 5-HT1A. Studi sperimentali nell’uomo hanno anche suggerito che meccanismi serotoninergici possano contribuire a effetti ansiolitici acuti in certe condizioni. Ma “contribuisce a” non significa “agisce principalmente attraverso”.
CBD è farmacologicamente ampio. Ha bassa affinità per CB1 e CB2 rispetto a THC, ma questo non lo rende automaticamente un agente serotoninergico a target singolo. Nella letteratura, i contributi plausibili includono TRPV1, 5-HT1A, GPR55, segnalazione dell’adenosina, PPAR-gamma, gestione del calcio intracellulare, tono endocannabinoid legato a FAAH in alcuni contesti e gli effetti dei metaboliti che possono non corrispondere al composto madre. La concentrazione conta qui più di quanto molti divulgatori ammettano. Un effetto recettoriale visto in vitro a livelli micromolari può non dominare nell’uomo dopo una dose orale standard, dove l’assorbimento è variabile e il metabolismo di primo passaggio è sostanziale.
Il record clinico si oppone alle tesi a un solo meccanismo. L’uso FDA più forte di CBD purificato non è l’ansia, ma i disturbi convulsivi: la soluzione orale è indicata per la sindrome di Lennox-Gastaut, la sindrome di Dravet e il complesso della sclerosi tuberosa in pazienti di età pari o superiore a 1 anno. Questo uso approvato dice già qualcosa. Se CBD fosse “principalmente serotonina”, il suo profilo terapeutico più riproducibile sarebbe difficile da conciliare con ciò che i clinici lo usano davvero per trattare. La serotonina può contare negli scenari legati all’ansia, soprattutto nelle risposte legate a 5-HT1A, ma la farmacologia umana di CBD non si riduce a un’etichetta serotoninergica.
Anche nell’ansia, il meccanismo probabilmente cambia con dose e contesto. TRPV1 è un buon esempio. CBD può attivare TRPV1, e la segnalazione TRP è così centrale per la biologia sensoriale che David Julius e Ardem Patapoutian hanno ricevuto il premio Nobel 2021 per le scoperte dei recettori per temperatura e tatto. Tuttavia, gli effetti di TRPV1 non sono lineari. L’attivazione può essere seguita da desensibilizzazione; dosi basse e alte possono produrre esiti comportamentali diversi. Quindi quando qualcuno dice “CBD agisce tramite la serotonina”, la risposta giusta è: in parte, a volte, e probabilmente non da sola.
Un solo target può spiegare l’effetto di una pianta intera
Nessun singolo target può spiegare l’effetto di una pianta intera di cannabis, e cercare di forzarlo di solito oscura più di quanto chiarisca.
Gli effetti della pianta intera emergono da variabili impilate. Iniziare dalla composizione: THC, CBD, minor cannabinoid come CBG, CBC, THCV, precursori acidi, prodotti di ossidazione e metaboliti portano tutti profili di target distinti. Aggiungere la via di somministrazione e il quadro cambia di nuovo. I cannabinoid inalati raggiungono il cervello rapidamente; i prodotti orali subiscono metabolismo di primo passaggio e generano specie attive diverse. Il diritto statunitense definisce ancora l’hemp con una soglia di delta-9 THC di “not more than 0.3 percent on a dry weight basis,” ma questa linea legale dice poco sulla farmacologia di tutto il resto del campione o su ciò che si forma dopo il metabolismo.
Poi si aggiunge la specificità tissutale. Un cannabinoid può influenzare parallelamente CB1 centrale, canali TRP periferici, segnalazione immunitaria e recettori nucleari. Il report 2025 della Hebrew University secondo cui THC inibisce i nocicettori periferici prendendo di mira NaV1.7 e NaV1.8 è un esempio netto perché rompe l’equazione pigra “effetto THC=effetto CB1.” Se persino THC ha azioni significative sui canali del sodio nelle vie del dolore, allora l’idea che un fiore, un estratto o un edibile abbia un solo recettore maestro diventa difficile da difendere. La ricerca del 2026 evidenziata da ScienceDaily—un composto del cannabis che allevia il dolore senza il high—va nella stessa direzione, anche se resta allo stadio di ricerca. L’analgesia può essere separabile dall’intossicazione centrale sfruttando restrizione periferica o target non CB1. È lì che il campo sta andando.
Anche l’aspettativa e la biologia individuale contano. Esperienza pregressa, livello di ansia, genetica, sesso, attività degli enzimi epatici, sonno, infiammazione e farmaci concomitanti spostano tutti ciò che un dato prodotto fa sentire e ciò che produce fisiologicamente. I terpeni possono contribuire in alcuni casi, ma non vanno trattati come direttori magici dell’effetto complessivo. Le loro concentrazioni sono spesso basse e le evidenze umane sono più deboli del linguaggio di marketing.
La visione più dura ma più accurata è questa: gli effetti del cannabis sono emergenti. Nascono da molte interazioni modeste, non da un recettore slogan. Questo rende la scienza meno ordinata. La rende anche più onesta.
Interpretazione pratica per lettori, clinici e ricercatori
La lezione pratica della farmacologia cannabinoid non CB1/CB2 è semplice ma esigente: le affermazioni sul meccanismo dovrebbero ricevere un esame più severo, non un’accettazione più facile, quando un articolo invoca canali TRP, PPAR, GPR55, 5-HT1A, segnalazione dell’adenosina o canali NaV. I cannabinoid sono spesso molecole farmacologicamente promiscue. Questo può essere utile nel drug discovery. Può anche fuorviare i lettori, inducendoli a trattare qualsiasi colpo recettoriale in una piastra come spiegazione di un effetto clinico.
Una buona regola è classificare le evidenze in base alla distanza dal paziente. Un saggio di legame è l’inizio, non la fine. Gli studi di segnalazione cellulare vengono dopo. Il lavoro animale può rafforzare la plausibilità. La farmacologia sperimentale umana conta di più. Le evidenze da farmaco approvato contano di più di tutte, e anche lì l’etichetta può non risolvere il meccanismo. La soluzione orale di cannabidiol, per esempio, è approvata dalla FDA per convulsioni associate alla sindrome di Lennox-Gastaut, alla sindrome di Dravet e al complesso della sclerosi tuberosa in pazienti di età pari o superiore a 1 anno, eppure il suo profilo terapeutico non è spiegato in modo pulito da CB1 o CB2. Quel divario è esattamente il motivo per cui le affermazioni su TRPV1, GPR55, 5-HT1A e target intracellulari continuano a riemergere.
Come leggere in modo critico le affermazioni sul meccanismo dei cannabinoid
Iniziare dalla specie e dal sistema. L’effetto è stato mostrato in tessuto umano, in una linea cellulare murina, in ovociti di Xenopus o in recettori sovraespressi in HEK293? Non sono intercambiabili. GPR55 è un esempio perfetto: uno studio può mostrare CBD comportarsi da antagonista in un sistema ricombinante, mentre un altro contesto produce una segnalazione debole o variabile perché espressione recettoriale, lipidi endogeni e progettazione del saggio differiscono. Chiamare questo “il meccanismo” è di solito prematuro.
Poi porre la domanda sulla concentrazione. È qui che molte affermazioni sui cannabinoid crollano. Un articolo può riportare attivazione di TRPV1, transattivazione di PPAR-gamma o inibizione dei canali del sodio a concentrazioni micromolari. Bene. Ma la dose umana discussa produce davvero concentrazioni tissutali libere in quell’intervallo? I cannabinoid orali affrontano metabolismo di primo passaggio, legame proteico esteso e distribuzione tissutale disomogenea. Un colpo al target a 30 micromolari in vitro può essere chimica interessante e farmacologia clinica irrilevante. Può accadere anche il contrario: accumulo tissutale locale, metaboliti attivi o partizione lipidica possono rendere un target intracellulare più plausibile di quanto suggeriscano i soli livelli plasmatici. In ogni caso, la concentrazione non è una questione secondaria. È la questione.
Conta anche la misurazione diretta del target. Gli investigatori hanno davvero bloccato l’effetto con un antagonista selettivo, ridotto l’espressione del recettore o misurato la corrente del canale? Oppure hanno inferito il meccanismo dalla somiglianza con la letteratura precedente? Per i canali TRP, soprattutto TRPV1 e TRPA1, questo conta perché l’attivazione può essere bifasica e desensibilizzante. Un composto può prima attivare un canale e poi ridurre la responsività a valle, il che significa che “agonista” non si traduce sempre in modo lineare in “più dolore” o “più sensazione di calore.” Questa è una delle ragioni per cui il premio Nobel 2021 a David Julius e Ardem Patapoutian, assegnato “for their discoveries of receptors for temperature and touch,” è così rilevante qui: la segnalazione somatosensoriale è meccanicisticamente ricca, e le etichette recettoriali semplicistiche spesso falliscono.
La storia dei NaV mostra come appaia un’evidenza più forte. I ricercatori della Hebrew University hanno riportato nel 2025 che THC inibisce i nocicettori periferici prendendo di mira i canali del sodio nocicettivi NaV1.7 e NaV1.8. Questa affermazione è più informativa di vaghe frasi secondo cui THC “works outside CB1,” perché nomina canali rilevanti per il dolore già centrali nella ricerca analgesica. Riformula anche una vecchia assunzione. Anche un composto famoso per l’intossicazione mediata da CB1 può avere azioni non CB clinicamente rilevanti, soprattutto nel tessuto periferico.
I lettori dovrebbero anche cercare spiegazioni concorrenti. Supponiamo che uno studio colleghi CBD all’ansiolisi tramite 5-HT1A. Ipotesi ragionevole. Ma è stata esclusa la sedazione? La modulazione di CB1 è indiretta? L’esperimento ha distinto gli effetti recettoriali dai cambiamenti nel tono endocannabinoid, nel reuptake dell’adenosina, nella segnalazione infiammatoria o nell’aspettativa nei soggetti umani? I farmaci multi-target raramente annunciano quale target stia facendo la maggior parte del lavoro in un dato modello.
L’attuale contesto regolatorio rende questo scetticismo più che accademico. Nel 2025, HHS ha affermato che “7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” sostenendo un’azione di scheduling sui prodotti 7-OH potenziati. Non è un caso cannabis, ma la lezione si applica chiaramente: i regolatori distinguono sempre più i costituenti vegetali familiari dagli intossicanti potenziati, semi-sintetici o comunque modificati con diversi profili di potenza e sicurezza. Una politica sui cannabinoid che tratti tutti i composti come versioni scalate di delta-9-THC è farmacologicamente superata.
Quali domande dovrebbero porsi i clinici sulla rilevanza del target
I clinici non devono memorizzare ogni GPCR orfano per interpretare bene le affermazioni sui cannabinoid. Devono avere una checklist disciplinata.
Primo: l’effetto è stato mostrato nell’uomo o solo in cellule e animali? Un modello infiammatorio murino può sostenere la plausibilità per PPAR-gamma o TRPA1, ma non stabilisce il beneficio per il paziente. Secondo: a quali concentrazioni o dosi? Se un target proposto è coinvolto solo a livelli superiori a quelli raggiunti dal dosaggio orale standard, quel meccanismo potrebbe non spiegare gli esiti clinici di routine. Terzo: il target è stato misurato direttamente? Occupazione recettoriale, elettrofisiologia, inversione con antagonista o perturbazione genetica contano più dell’inferenza narrativa.
Quarto: la via di somministrazione rende la tesi più o meno credibile? Inalazione, assunzione orale, somministrazione transdermica e applicazione topica creano profili di esposizione molto diversi. Un meccanismo analgesico periferico è più plausibile per un composto topico o perifericamente ristretto che per una molecola che inonda rapidamente il cervello. Questa distinzione conta se l’obiettivo terapeutico è analgesia senza intossicazione.
Quinto: esistono spiegazioni concorrenti legate agli effetti avversi noti? Se un paziente riferisce meno ansia dopo una preparazione cannabinoid, si tratta di un effetto ansiolitico mediato da 5-HT1A, di minore dolore, di sedazione aspecifica o di aspettativa? Se i marcatori infiammatori cambiano, PPAR-gamma è il probabile motore oppure sono avvenuti cambiamenti metabolici o immunitari più ampi a monte? Il meccanismo non dovrebbe essere inferito solo dal miglioramento dei sintomi.
Per i clinici, anche i minor cannabinoid meritano la stessa cautela. CBC, CBG, THCV e i cannabinoid acidi vengono spesso discussi come se ciascuno avesse una firma stabile di target. La letteratura non giustifica ancora questa sicurezza. Alcuni sono promettenti. Nessuno dovrebbe essere considerato farmacologicamente definito solo perché un marchio o un thread social ne rivendica la specificità recettoriale.
Dove probabilmente andrà il campo nei prossimi anni
La direzione a breve più credibile è l’analgesia periferica. L’attrattiva traslazionale è evidente: separare il sollievo dal dolore dall’intossicazione centrale. Il report 2026 di ScienceDaily che descrive lavori su “a cannabis compound that relieves pain without the high” dovrebbe essere letto come un segnale allo stadio di ricerca, non come una risposta clinica definitiva, ma cattura bene dove punta la chimica medicinale. Lo stesso vale per il lavoro 2025 della Hebrew University su NaV1.7/NaV1.8: la biologia del dolore sta spingendo la scienza cannabinoid verso nervi periferici, canali ionici ed esposizione selettiva nei tessuti.
I ligandi nucleari anti-infiammatori sono un’altra strada forte. Le affermazioni su PPAR-gamma necessitano di dati migliori sull’impegno del target nell’uomo, ma il concetto è abbastanza plausibile da giustificare uno sviluppo serio, soprattutto dove vie metaboliche e infiammatorie si intersecano. È probabile che restino attive anche molecole ansiolitiche multi-target. MIRA Pharmaceuticals ha affermato in un comunicato Nasdaq del 2025 che il proprio candidato MIRA-55 mostrava un “differentiated mechanism of action” e “anxiolytic activity relative to THC” nei dati preclinici. Poiché si tratta di affermazione aziendale e preclinica, va trattata con cautela. Tuttavia, riflette un trend reale: i ricercatori non sono più soddisfatti di chiedersi se un candidato sia “simile a THC” o “simile a CBD.” Vogliono profili di target definiti.
Questo stesso spostamento è visibile nella chimica medicinale. L’articolo del 2016 del Journal of Medicinal Chemistry “Library Docking for Cannabinoid-2 Receptor Ligands” ha segnalato un approccio basato sulla struttura che da allora non ha fatto che crescere: costruire ligandi per target specifici, tessuti specifici e bias di segnalazione specifici. I minor cannabinoid meglio definiti emergeranno probabilmente da questa mentalità, non da affermazioni ampie secondo cui ogni cannabinoid raro avrebbe una nicchia unica nel benessere.
L’insegnamento pratico è netto. Quando si legge la farmacologia dei cannabinoid, porre ogni volta cinque domande: l’effetto è stato mostrato nell’uomo o solo nelle cellule? A quali concentrazioni? Il target è stato misurato direttamente? La dose raggiunge quel target nel tessuto vivente? Esistono spiegazioni concorrenti? Il futuro della scienza cannabinoid non è una farmacologia meno specifica, ma una più specifica.
Riferimenti
- [1]HHS and FDA support DEA action on dangerous 7-OH products. HHS Press Room, 2025. https://www.hhs.gov/press-room/hhs-fda-support-dea-7-oh-scheduling.html
- [2]EPIDIOLEX (cannabidiol) oral solution label. FDA drug label, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/210365s016lbl.pdf
- [3]Library Docking for Cannabinoid-2 Receptor Ligands. Journal of Medicinal Chemistry, 2016. https://pubs.acs.org/doi/10.1021/acs.jmedchem.6c00835
- [4]MIRA Pharmaceuticals Reports New Preclinical Data Demonstrating MIRA-55's Differentiated Mechanism of Action and Anxiolytic Activity Relative to THC. Nasdaq press release, 2025. https://www.nasdaq.com/press-release/mira-pharmaceuticals-reports-new-preclinical-data-demonstrating-mira-55s
- [5]Psychoactive cannabinoid THC inhibits peripheral nociceptors by targeting NaV1.7 and NaV1.8 nociceptive sodium channels. Research portal summary, 2025. https://cannabinoids.huji.ac.il/publications/psychoactive-cannabinoid-thc-inhibits-peripheral-nociceptors-targeting
- [6]A cannabis compound that relieves pain without the high. ScienceDaily, 2026. https://www.sciencedaily.com/releases/2026/06/260619033343.htm
- [7]The Nobel Prize in Physiology or Medicine 2021. Nobel Prize Press Release, 2021. https://www.nobelprize.org/prizes/medicine/2021/press-release/
- [8]Agriculture Improvement Act of 2018. Congress.gov, 2018. https://www.congress.gov/bill/115th-congress/house-bill/2/text