






Inhaltsverzeichnis
- Warum Cannabinoid-Wissenschaft nicht auf CB1 und CB2 reduziert werden kann
- Das Endocannabinoid-System versus die breitere Cannabinoid-Ziellandschaft
- TRP-Kanäle: die Hitze-, Schmerz- und Reizsensoren, die Cannabinoide ständig treffen
- PPARs: Cannabinoide als intrazelluläre Lipidsignale, nicht nur als Liganden von Membranrezeptoren
- GPR55, GPR18 und GPR119 und das Orphan-GPCR-Problem
- Serotonin-Signalgebung: wo Cannabinoide mit 5-HT-Systemen zusammentreffen
- Jenseits der angeforderten Liste: Natriumkanäle und andere nichtkanonische Zielstrukturen, die die Schmerzdebatte bereits verändern
- Wie sich spezifische Cannabinoide unterscheiden, wenn man nicht mehr nur nach CB1 und CB2 fragt
- Methoden sind entscheidend: warum das Assay-Design prägt, was wir über die Wirkung von Cannabinoiden glauben
- Evidenzstufen: von der Zellschale bis zur Klinik
- Sicherheit, Regulierung und warum Off-Target-Pharmakologie für die öffentliche Gesundheit wichtig ist
- Arzneimittelforschung: Cannabinoide und cannabinoid-inspirierte Moleküle für Nicht-CB1/CB2-Zielstrukturen designen
- Häufige Missverständnisse und ungelöste Kontroversen
- Praktische Einordnung für Leser, Kliniker und Forscher
Warum Cannabinoid-Wissenschaft nicht auf CB1 und CB2 reduziert werden kann
Die Kurzfassung der Cannabinoid-Pharmakologie lautet: THC wirkt an CB1, immunologische Effekte laufen über CB2, und alles andere ist eine Fußnote. Diese Darstellung ist leicht zu lehren und leicht zu wiederholen. Sie ist jedoch oft genug falsch, um ein ernsthaftes Verständnis von Schmerz, Entzündung, Angst, Juckreiz, Übelkeit, Stoffwechsel und Neuroprotektion zu behindern.
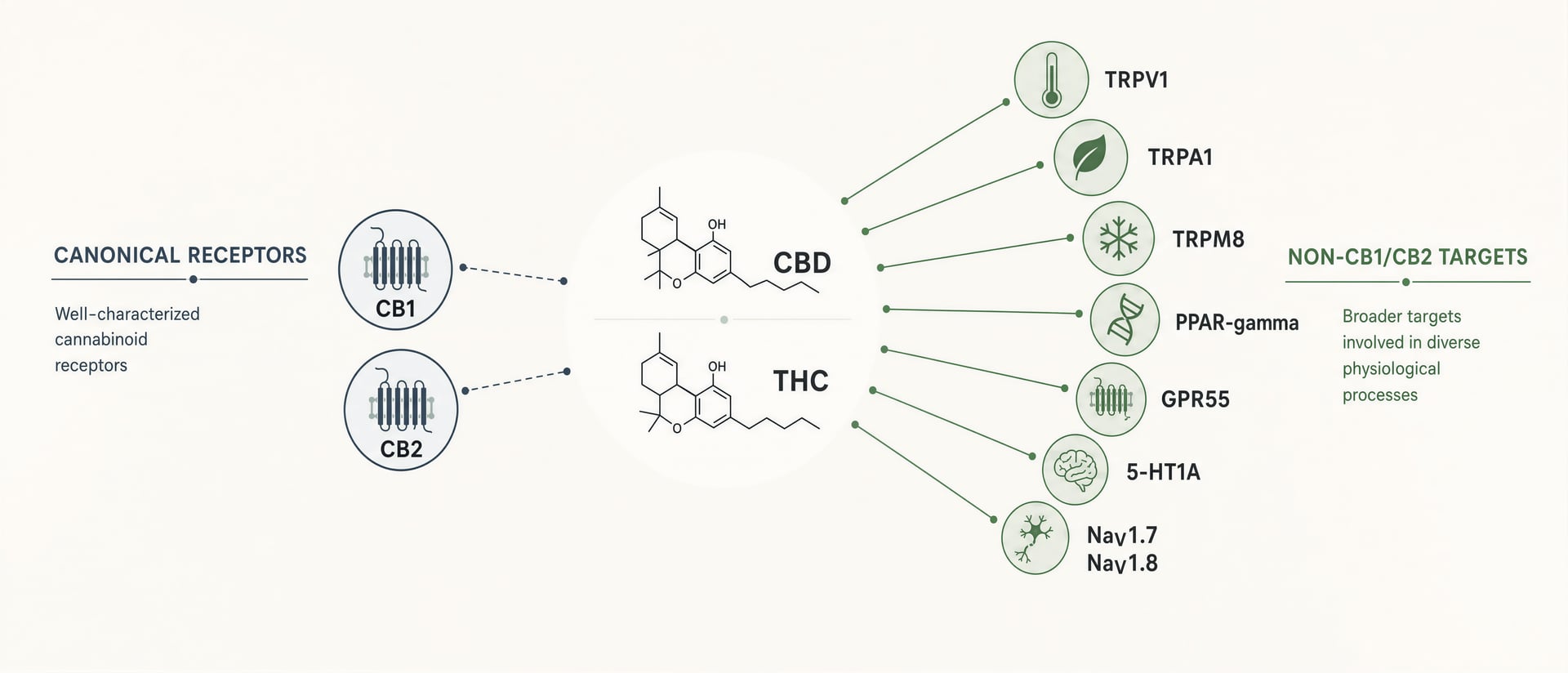
CB1 und CB2 sind wichtig. CB1 ist im Gehirn reichlich vorhanden und erklärt einen Großteil der Intoxikation, der Gedächtnisbeeinträchtigung, der appetitfördernden Effekte und eines Teils der Analgesie von THC. CB2 steht im Zentrum vieler immunologischer und entzündlicher Diskussionen. Aber Cannabinoide sind keine ordentlichen Liganden, die jeweils für einen einzigen Rezeptor gebaut wurden. Es handelt sich um lipophile, strukturell flexible Moleküle, die mit einem breiteren pharmakologischen Feld interagieren: Transiente Rezeptorpotential-Kanäle wie TRPV1 und TRPA1, nukleäre Rezeptoren wie PPAR-gamma, Orphan- oder noch umstrittene cannabinoidnahe GPCRs wie GPR55 und GPR18, Serotoninrezeptoren einschließlich 5-HT1A, adenosinbezogene Signalwege, den Fett- und Fettsäuretransport sowie den Stoffwechsel und, in neueren Arbeiten, spannungsabhängige Natriumkanäle einschließlich NaV1.7 und NaV1.8.[1]HHS and FDA support DEA action on dangerous 7-OH products. U.S. Department of Health and Human Services. HHS Press Room, 2025. https://www.hhs.gov/press-room/hhs-fda-support-dea-7-oh-scheduling.html
Dieses breitere Feld ist wichtig, weil der Mechanismus über Risiko und Nutzen entscheidet. Regulierungsbehörden sehen sich dieses Problems bereits in angrenzenden drogenpolitischen Auseinandersetzungen gegenüber. Im Jahr 2025 erklärte das U.S. Department of Health and Human Services, dass „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety“ sei, als es eine DEA-Maßnahme gegen verstärkte 7-OH-Produkte unterstützte. Die konkrete Verbindung ist kein Cannabinoid, aber die politisch-pharmakologische Lehre ist übertragbar: Sobald Chemiker beginnen, natürliche Gerüststrukturen zu modifizieren und Metaboliten zu konzentrieren, schützen einfache, quellenbasierte Kategorien die Öffentlichkeit nicht mehr. Das Zielprofil eines Moleküls ist wichtiger als die Tatsache, dass populäre Texte es als vertraut darstellen.
Der Rezeptor-Mythos in populären Cannabis-Texten
Populäre Cannabis-Erklärungen stellen Rezeptoren gewöhnlich als Ein-/Aus-Schalter dar: THC aktiviert CB1, CBD bindet nicht „stark“, also müsse CBD schwach oder mysteriös sein. Diese Darstellung vermengt mehrere unterschiedliche pharmakologische Konzepte in einem vagen Verb: binden.
Orthosterische Agonistik ist der klassische Fall. Ein Ligand besetzt die Hauptbindungsstelle des Rezeptors und stabilisiert die Signalübertragung. THC ist ein partieller Agonist an CB1 und CB2. Das ist eine Wirkungsweise, aber nicht das Muster der gesamten Cannabinoid-Biologie. Eine Verbindung kann stattdessen allosterisch wirken und verändern, wie ein anderer Ligand am Rezeptor arbeitet, ohne dieselbe Stelle zu besetzen. Sie kann einen Ionenkanal öffnen, sensibilisieren oder desensibilisieren. Sie kann in die Zelle eintreten und einen nukleären Rezeptor aktivieren, der die Genexpression über Stunden statt Millisekunden verändert. Sie kann einen Transporter hemmen, Membraneigenschaften verändern oder ein Enzym verlangsamen, das ein endogenes Signallipid abbaut.[2]EPIDIOLEX (cannabidiol) oral solution label. U.S. Food and Drug Administration. FDA drug label, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/210365s016lbl.pdf
CBD ist die klarste Widerlegung des Rezeptor-Reduktionismus. Seine zugelassene klinische Anwendung beruht nicht auf CB1-Agonismus. Die FDA-Kennzeichnung für Cannabidiol-Orallösung gibt an, dass sie zur Behandlung von Anfällen im Zusammenhang mit dem Lennox-Gastaut-Syndrom, dem Dravet-Syndrom und dem tuberösen Sklerose-Komplex bei Patienten ab 1 Jahr indiziert ist. Welcher vollständige Mechanismus diesem Effekt auch zugrunde liegen mag, er wird durch die alte Erzählung nicht angemessen erklärt, wonach relevante Cannabinoid-Wirkung gleichbedeutend mit starker CB1- oder CB2-Aktivierung sei. Zu den mechanistischen Kandidaten, die in der Literatur wiederholt genannt werden, gehören TRPV1, 5-HT1A-bezogene Signalgebung, Adenosin-Modulation, intrazelluläre Calcium-Effekte sowie Enzym- oder Transporterinteraktionen. Keiner kann als alleinige Antwort behandelt werden, doch zusammen zeigen sie, warum der vereinfachende Rezeptor-Mythos scheitert.
Auch die Geschichte weist in dieselbe Richtung. Die Arbeit von Raphael Mechoulam zum Endocannabinoid-System eröffnete ein Feld, das sich auf Anandamid und 2-AG konzentrierte, doch selbst Anandamid ist nicht bloß ein CB1-Ligand. Es aktiviert auch TRPV1, den Hitze- und Capsaicin-Rezeptor, dessen breitere sensorische Bedeutung durch den Nobelpreis 2021 an David Julius und Ardem Patapoutian für Entdeckungen von Rezeptoren für Temperatur und Berührung anerkannt wurde. Sobald ein endogenes Cannabinoid sowohl über einen GPCR als auch über einen TRP-Kanal signalisieren kann, ist das Modell „nur CB1/CB2“ kein Modell mehr. Es ist eine Karikatur.
Polypharmakologie: ein Ligand, viele Ziele
Ein besserer Ausgangspunkt ist Polypharmakologie. Ein Ligand, viele Ziele, mit unterschiedlichen Affinitäten, Wirksamkeiten, Geweben und Konsequenzen. In der Pharmakologie wird „dirty“ manchmal abwertend verwendet, bei Cannabinoiden ist es jedoch häufig schlicht beschreibend.
Betrachten Sie, wie viele Wirkungsarten unter demselben Oberbegriff zusammengefasst werden. THC ist ein CB1/CB2-partieller Agonist, doch 2025 hob von der Hebrew University hervorgehobene Arbeit hervor, dass THC periphere Nozizeptoren hemmt, indem es auf die nozizeptiven Natriumkanäle NaV1.7 und NaV1.8 abzielt. Das ist überhaupt keine Rezeptor-Agonistik. Es ist eine Ionenkanalhemmung an Zielstrukturen, die bereits als vielversprechende Schmerzmedikamenten-Ziele gelten. Wenn sich diese Arbeit über Spezies und Dosierungsbedingungen hinweg bestätigt, könnte ein Teil der analgetischen Wirkung von THC auf einem Mechanismus beruhen, der eher wie eine lokale Erregbarkeitsbremse als wie ein klassischer Cannabinoid-Rezeptor-Effekt aussieht.
CBD zeigt eine andere Art von Promiskuität. Über Assaysysteme hinweg wurde berichtet, dass es unter anderem TRPV1, TRPA1, TRPM8, 5-HT1A, PPAR-gamma, GPR55 und den Adenosin-Tonus beeinflusst. Das Problem ist nicht ein Mangel an Mechanismen. Das Problem besteht darin, zu sortieren, welche Mechanismen bei Konzentrationen relevant sind, die beim Menschen tatsächlich erreicht werden. In-vitro-Target-Bindung ist billig. Die Translation ist schwierig. Ein Mikromolar-Effekt in einer überexprimierenden Zelllinie erklärt nicht automatisch Patientenbefunde nach oraler Gabe, First-Pass-Metabolismus, Proteinbindung und Gewebeverteilung.
Andere Phytocannabinoide verkomplizieren das Bild weiter. CBG wurde in einigen Systemen als alpha-2-adrenerges, TRP-aktives und mit 5-HT1A interagierendes Molekül diskutiert. CBC wurde mit TRPA1- und TRPV-Kanälen in Verbindung gebracht. THCV kann sich je nach Dosis und Kontext an CB1 anders verhalten als Delta-9-THC und zugleich nicht-CB1-Optionen mitbringen. Saure Cannabinoide wie CBDA und THCA werfen zusätzliche Fragen auf, weil Decarboxylierung, Stabilität und Metabolitenbildung die Zielstruktur-Exposition verändern. Dasselbe Fläschchen kann daher je nach Applikationsweg, Hitze, Metabolismus und Formulierung eine sehr unterschiedliche Pharmakologie verbergen.[3]Library Docking for Cannabinoid-2 Receptor Ligands. American Chemical Society. Journal of Medicinal Chemistry, 2016. https://pubs.acs.org/doi/10.1021/acs.jmedchem.6c00835
Auch innerhalb der GPCR-Pharmakologie hat sich das Feld von groben Bezeichnungen entfernt. GPR55 wird gelegentlich noch als „CB3“-Kandidat bezeichnet, doch das bleibt aus gutem Grund umstritten; Signalgebung, Ligandenspektrum und physiologische Rolle passen nicht sauber zu den klassischen Cannabinoid-Rezeptoren. GPR18 und GPR119 werden ebenfalls in cannabinoidnaher Literatur diskutiert, insbesondere im Zusammenhang mit Entzündung, Stoffwechsel und Darm-Signalisierung, aber die Evidenz ist uneinheitlich. Das wissen auch Medizinalchemiker. Ein Paper aus dem Jahr 2016 im Journal of Medicinal Chemistry, „Library Docking for Cannabinoid-2 Receptor Ligands“, steht für einen struktur-basierten Ansatz, der fast das Gegenteil der populären Rezeptor-Erzählung ist: zielselektives Design, Docking, Gerüstoptimierung und bewusste Trennung gewünschter von unerwünschten Effekten. Das Feld fragt nicht „trifft es Cannabinoid-Rezeptoren?“, sondern welche Ziele, in welchem Zustand, in welchem Gewebe, bei welcher Konzentration und mit welcher Signal-Bias.
Warum nicht-CB1/CB2-Ziele klinisch wichtig sind
Hier hört die Wissenschaft auf, semantisch zu sein, und beginnt, Medizin zu beeinflussen.
Bei Schmerz könnten Nicht-CB1-Ziele der plausibelste Weg zu nützlichen Arzneimitteln mit weniger Intoxikation sein. TRPV1, TRPA1, periphere Natriumkanäle und entzündliche Transkriptionswege bieten Möglichkeiten, Nozizeptor-Feuerung oder neuroimmune Sensibilisierung zu senken, ohne starke zentrale CB1-Aktivierung. Ein 2026-ScienceDaily-Bericht über eine Cannabis-Verbindung, die „pain without the high“ lindert, ist nur ein Forschungssignal, keine fertige klinische Antwort, aber die Richtung ist sinnvoll. Wenn Analgesie auf periphere Ionenkanäle oder eine auf Gewebe begrenzte Exposition verschoben werden kann, könnte sich der alte Kompromiss zwischen Schmerzlinderung und psychoaktiver Belastung abschwächen.
Bei Entzündung und Stoffwechsel ist PPAR-gamma ein gutes Beispiel dafür, warum Rezeptorkategorien wichtig sind. PPARs sind nukleäre Rezeptoren, keine membranständigen Cannabinoid-Rezeptoren. Ihre Aktivierung verändert Genexpressionsprogramme, die Lipidverarbeitung, Insulinsensitivität und entzündlichen Tonus betreffen. Einige Cannabinoid-Effekte, die in metabolischen oder entzündlichen Modellen berichtet werden, passen besser zu dieser langsameren Transkriptionsbiologie als zu schneller CB1-Signalgebung. Aber auch hier gilt: Konzentration und intrazellulärer Zugang sind entscheidend. Ein Reporter-Assay, der PPAR-Aktivierung zeigt, beweist keinen klinisch relevanten antiinflammatorischen Effekt beim Menschen.[4]MIRA Pharmaceuticals Reports New Preclinical Data Demonstrating MIRA-55's Differentiated Mechanism of Action and Anxiolytic Activity Relative to THC. MIRA Pharmaceuticals. Nasdaq press release, 2025. https://www.nasdaq.com/press-release/mira-pharmaceuticals-reports-new-preclinical-data-demonstrating-mira-55s
Bei Angst und Übelkeit tauchen serotonerge Mechanismen immer wieder auf, insbesondere 5-HT1A. Die Daten sind gemischt und oft indirekt, aber die Persistenz des Signals ist aufschlussreich. Die anxiolytische Reputation von CBD ist schwer allein auf CB1/CB2 zu beziehen. Das ist ein Grund, warum Unternehmen versuchen, differenzierte cannabinoid-inspirierte Verbindungen zu entwickeln, anstatt einfach stärkere THC-Analoga zu bauen. Im Jahr 2025 meldete MIRA Pharmaceuticals präklinische Daten, wonach der Kandidat MIRA-55 einen „differentiated mechanism of action“ und „anxiolytic activity relative to THC“ gezeigt habe. Pressemitteilungen von Unternehmen liefern Evidenz niedriger Stufe und sollten auch so behandelt werden. Dennoch zeigen sie, wohin die Arzneimittelentwicklung geht: weg von der Idee, dass die beste Cannabinoid-Medizin nur eine sauberere CB1-Stimulation sei.
Juckreiz, Migräne, Epilepsie, Darmerkrankungen und Neuroprotektion liegen alle in derselben mechanistischen Zone. TRP-Kanäle regulieren die sensorische Verstärkung. GPRs können immunologische und epitheliale Signalgebung prägen. PPARs verändern Entzündungsprogramme. Natriumkanäle kontrollieren direkt die Erregbarkeit. Serotoninwege beeinflussen Angst, Emesis und Stressreaktionen. Sobald diese Systeme neben CB1 und CB2 gestellt werden statt unter sie, wirken viele reale Cannabinoid-Effekte weniger mysteriös und pharmakologisch gewöhnlicher.
Das vereinfachte Modell überlebt, weil es leicht ist. Das bessere Modell überlebt den Datenkontakt.
Das Endocannabinoid-System versus die breitere Cannabinoid-Ziellandschaft
Populäre Cannabis-Texte behandeln Pharmakologie oft als Zwei-Rezeptor-Geschichte: CB1 erklärt psychoaktive Effekte, CB2 erklärt immunologische Effekte, und alles andere ist Detail. Diese Darstellung ist für die Evidenz zu klein. Sie übersieht, warum Cannabidiol nicht sauber über CB1 oder CB2 erklärt werden kann, warum manche Cannabinoide über TRP-Kanäle Brennen oder Analgesie auslösen, warum intrazelluläre nukleäre Rezeptoren wie PPAR-gamma in Entzündungsstudien ständig auftauchen und warum sogar THC selbst schmerzrelevante Natriumkanäle außerhalb der klassischen Cannabinoid-Signalgebung beeinflussen kann. Wenn das Feld Schmerz, Angst, Entzündung, Krampfkontrolle oder Sicherheitsprobleme mit neuartigen Intoxikanzien erklären will, muss der Rezeptor-Reduktionismus weichen.
Der regulatorische Moment macht das deutlich. 2025 erklärte HHS, dass „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety“ sei, als es eine Einstufungsmaßnahme gegen verstärkte 7-OH-Produkte unterstützte. Diese Aussage bezog sich nicht auf cannabis, sie erfasst aber dieselbe pharmakologische Lehre: Sobald Hersteller von vertrauten Pflanzenbestandteilen zu verstärkten, halbsynthetischen oder strukturell modifizierten Intoxikanzien übergehen, verlieren einfache Kategorielabels ihren Nutzen. „THC-like“ sagt weit weniger aus als Zielprofil, Potenz, Metaboliten, Gewebeverteilung und Off-Target-Aktivität.
Kanonische Ziele: CB1, CB2, Anandamid und 2-AG
Das kanonische Endocannabinoid-System bleibt wichtig. CB1 und CB2 sind G-Protein-gekoppelte Rezeptoren, hauptsächlich Gi/o-gekoppelt, die Ende des 20. Jahrhunderts identifiziert und von Forschern einschließlich Ken Mackie und Vincenzo Di Marzo detailliert kartiert wurden. CB1 wird im zentralen Nervensystem stark exprimiert, insbesondere in Kortex, Hippocampus, Basalganglien und Kleinhirn, weshalb die partielle Agonistik von THC dort mit Intoxikation, Gedächtniseffekten, veränderter Motorik und Appetitveränderungen verknüpft ist. CB2 ist in Immunzellen und peripheren Geweben angereichert, wenn auch nicht im Gehirn abwesend. Die Aktivierung beider Rezeptoren reduziert gewöhnlich die cAMP-Bildung, moduliert Ionenkanäle und verändert die Transmitterfreisetzung.
Die endogenen Liganden sind Anandamid und 2-Arachidonoylglycerol, meist kurz Anandamid und 2-AG genannt. Die Gruppe um Raphael Mechoulam war zentral in dieser Geschichte: Anandamid wurde 1992 identifiziert, 2-AG kurz darauf. Sie werden nicht wie klassische Neurotransmitter in synaptischen Vesikeln gespeichert. Sie werden bei Bedarf aus Membranlipid-Vorstufen synthetisiert und wirken oft retrograd, indem sie von postsynaptischen Zellen zurück zu präsynaptischen Endigungen gelangen, um die Neurotransmitterfreisetzung zu dämpfen. Anandamid wird hauptsächlich durch FAAH abgebaut; 2-AG hauptsächlich durch MAGL. Dieser biochemische Kreislauf bildet das Rückgrat des Endocannabinoid-Systems.
Aber das Rückgrat ist nicht das ganze Skelett. Anandamid ist auch ein TRPV1-Agonist. CBD hat im Vergleich zu THC nur geringe direkte Affinität zu CB1 und CB2, zeigt jedoch eindeutig klinisch relevante Wirkungen; die von der FDA zugelassene orale Cannabidiol-Lösung ist bei Anfällen im Zusammenhang mit dem Lennox-Gastaut-Syndrom, dem Dravet-Syndrom und dem tuberösen Sklerose-Komplex bei Patienten ab 1 Jahr indiziert. Diese zugelassene Anwendung erinnert eindrücklich daran, dass klinisch relevante Cannabinoid-Effekte nicht zwangsläufig mit starker CB1-Agonistik übereinstimmen müssen.
Was als Cannabinoid-Ziel gilt
Eine pragmatische Definition ist besser als eine puristische. Ein Cannabinoid-Ziel ist jede molekulare Stelle, an der ein Phytocannabinoid, Endocannabinoid, Metabolit oder cannabinoid-inspiriertes Gerüst bei Konzentrationen, die in Zellen, Geweben, Tieren oder Menschen relevant sein können, bindet oder die Signalgebung funktionell moduliert. Nach dieser Definition weitet sich die Landschaft schnell aus.
TRP-Kanäle sind die bekanntesten Nicht-CB-Beispiele. TRPV1, TRPA1, TRPV2 und TRPM8 tauchen in Cannabinoid-Arbeiten immer wieder auf. Das ist keine Randnotiz. David Julius und Ardem Patapoutian erhielten 2021 den Nobelpreis für Physiologie oder Medizin „for their discoveries of receptors for temperature and touch“, eine Erinnerung daran, dass Ionenkanäle für Hitze, Kälte, Reizung und Mechanosensation direkt in Schmerzbahnen liegen. Anandamid aktiviert TRPV1. CBD, CBG, CBC und saure Cannabinoide haben in vitro alle TRP-Aktivität gezeigt, oft konzentrationsabhängig und manchmal biphasisch. Ein Cannabinoid, das TRPV1 zunächst aktiviert, kann es später desensibilisieren und so den Paradox erzeugen, dass anfängliche Reizung in Analgesie übergeht.
PPARs erweitern den Rahmen weiter. PPAR-α und PPAR-gamma sind nukleäre Rezeptoren, die Transkription in Bezug auf Lipidstoffwechsel und Entzündung regulieren. Einige Cannabinoide und endocannabinoidbezogene Lipide wirken hier direkt oder nach intrazellulärer Anreicherung und Metabolisierung. Das sind langsamere, genregulatorische Effekte, nicht die Millisekunden-Signalgebung von CB1. Das ist bedeutsam für chronische Entzündungsansprüche, die über nukleare Signalgebung oft besser erklärt werden als über akute synaptische Cannabinoid-Rezeptor-Aktivität.
Dann gibt es die Orphan- oder noch umstrittenen GPCRs, insbesondere GPR55, GPR18 und GPR119. GPR55 wurde wiederholt als „CB3“-Kandidat vorgeschlagen, und diese Bezeichnung ist verfrüht. Der Rezeptor ist real; die Klassifikation ist umstritten. CBD wird in experimentellen Systemen häufig als GPR55-Antagonist oder negativer Modulator beschrieben, während bestimmte endogene Lipide und synthetische Liganden ihn aktivieren können. GPR18 und GPR119 tauchen im Zusammenhang mit Entzündung, Stoffwechsel und Immunsignalisierung auf, doch die Evidenz ist uneinheitlich und Spezieseffekte können erheblich sein.
Serotoninrezeptoren, insbesondere 5-HT1A, gehören ebenfalls auf diese breitere Karte. Die anxiolytische und antiemetische Literatur zu CBD impliziert häufig 5-HT1A, auch wenn noch darüber gestritten wird, ob direkte Agonistik oder indirekte Förderung vorliegt. Diese Unterscheidung ist wichtig. Eine Verbindung, die schwach an einen Rezeptor bindet, aber die Schaltkreisfunktion zuverlässig über allosterische oder Netzwerkmechanismen verschiebt, kann in vivo dennoch bedeutsame Effekte haben. Dieselbe Vorsicht gilt für von Unternehmen berichtete präklinische Programme: 2025 sagte MIRA Pharmaceuticals, sein Kandidat MIRA-55 habe einen „differentiated mechanism of action“ und anxiolytische Aktivität im Vergleich zu THC gezeigt. Das ist noch kein Beleg für klinischen Nutzen, zeigt aber, wohin die Medizinalchemie geht – weg von grober THC-Nachahmung und hin zu zielgeformter Cannabinoid-Pharmakologie.[5]Psychoactive cannabinoid THC inhibits peripheral nociceptors by targeting NaV1.7 and NaV1.8 nociceptive sodium channels. Hebrew University of Jerusalem cannabinoids research portal. Research portal summary, 2025. https://cannabinoids.huji.ac.il/publications/psychoactive-cannabinoid-thc-inhibits-peripheral-nociceptors-targeting[6]A cannabis compound that relieves pain without the high. ScienceDaily. ScienceDaily, 2026. https://www.sciencedaily.com/releases/2026/06/260619033343.htm
Auch Natriumkanäle verdienen hier einen Platz. Ein Bericht der Hebrew University aus dem Jahr 2025 identifizierte THC-Hemmung peripherer Nozizeptoren über NaV1.7- und NaV1.8-nozizeptive Natriumkanäle. Das ist ein ernstzunehmender Befund, weil NaV1.7 und NaV1.8 zentrale Schmerzziele sind und der Mechanismus außerhalb von CB1/CB2 liegt. Er passt auch zu einem größeren translationalen Vorstoß. 2026 hob ScienceDaily Forschung zu „a cannabis compound that relieves pain without the high“ hervor. Der genaue Wirkstoff und die klinischen Aussichten müssen sorgfältig geprüft werden, aber die Richtung ist glaubwürdig: Analgesie kann sich zumindest prinzipiell von zentraler Intoxikation trennen lassen, indem periphere oder Nicht-CB1-Wege adressiert werden.
Signalverzerrung Eine Eigenschaft eines Liganden, bei der er Rezeptorzustände stabilisiert, die einen nachgeschalteten Signalweg gegenüber einem anderen begünstigen, wie etwa G-Protein-Signalisierung gegenüber beta-arrestin-Rekrutierung.
Affinität, Wirksamkeit, Bias und Konzentrationsfenster
Diese breitere Zielkarte ergibt nur Sinn, wenn die pharmakologischen Begriffe klar sind. Ki ist eine Bindungsaffinitätskonstante: Ein niedriger Ki-Wert bedeutet normalerweise stärkere Bindung in einem Konkurrenzassay. EC50 ist die Konzentration, die 50 Prozent eines gemessenen funktionellen Effekts auslöst. Beides ist nicht austauschbar. Ein Ligand kann stark binden, aber schwache Signalgebung erzeugen, oder mäßig binden und dennoch die Funktion durch Verstärkung in einem Signalweg stark verschieben.
Ein Agonist aktiviert einen Rezeptor. Ein Antagonist blockiert die Aktivierung durch einen anderen Liganden. Ein Inverse-Agonist verschiebt konstitutiv aktive Rezeptoren in Richtung niedrigerer Basissignalgebung. THC wird an CB1 gewöhnlich als partieller Agonist beschrieben: Selbst wenn es Rezeptoren besetzt, erzeugt es nicht den vollen Effekt eines hochwirksamen Agonisten. Das hilft zu erklären, warum verschiedene Cannabinoide und selbst verschiedene synthetische CB1-Liganden sehr unterschiedliche physiologische Obergrenzen haben können.
Signal-Bias bedeutet, dass ein Ligand Rezeptorkonformationen stabilisiert, die einen Signalweg gegenüber einem anderen bevorzugen, etwa G-Protein-Signalisierung gegenüber β-Arrestin-Rekrutierung. Das ist inzwischen Standard in der Arzneimittelentwicklung, auch in der cannabinoidbezogenen Medizinalchemie; das 2016er Journal of Medicinal Chemistry-Paper „Library Docking for Cannabinoid-2 Receptor Ligands“ steht in dieser gezielt entwickelten Tradition. Desensibilisierung bedeutet, dass wiederholte oder anhaltende Aktivierung die Reaktionsfähigkeit verringern kann, ein zentrales Problem bei TRP-Kanälen und bei CB1 selbst. Schließlich bedeutet gewebespezifische Target-Engagement, dass dieselbe Verbindung je nach Konzentration, Applikationsweg, Metabolismus und lokaler Proteinexpression unterschiedliche Ziele in Gehirn, Darm, Haut, Immunzellen oder peripheren Nerven trifft. Deshalb ist in-vitro-Promiskuität nicht automatisch klinische Relevanz – aber auch der Grund, warum CB1/CB2-only-Erklärungen immer wieder scheitern.
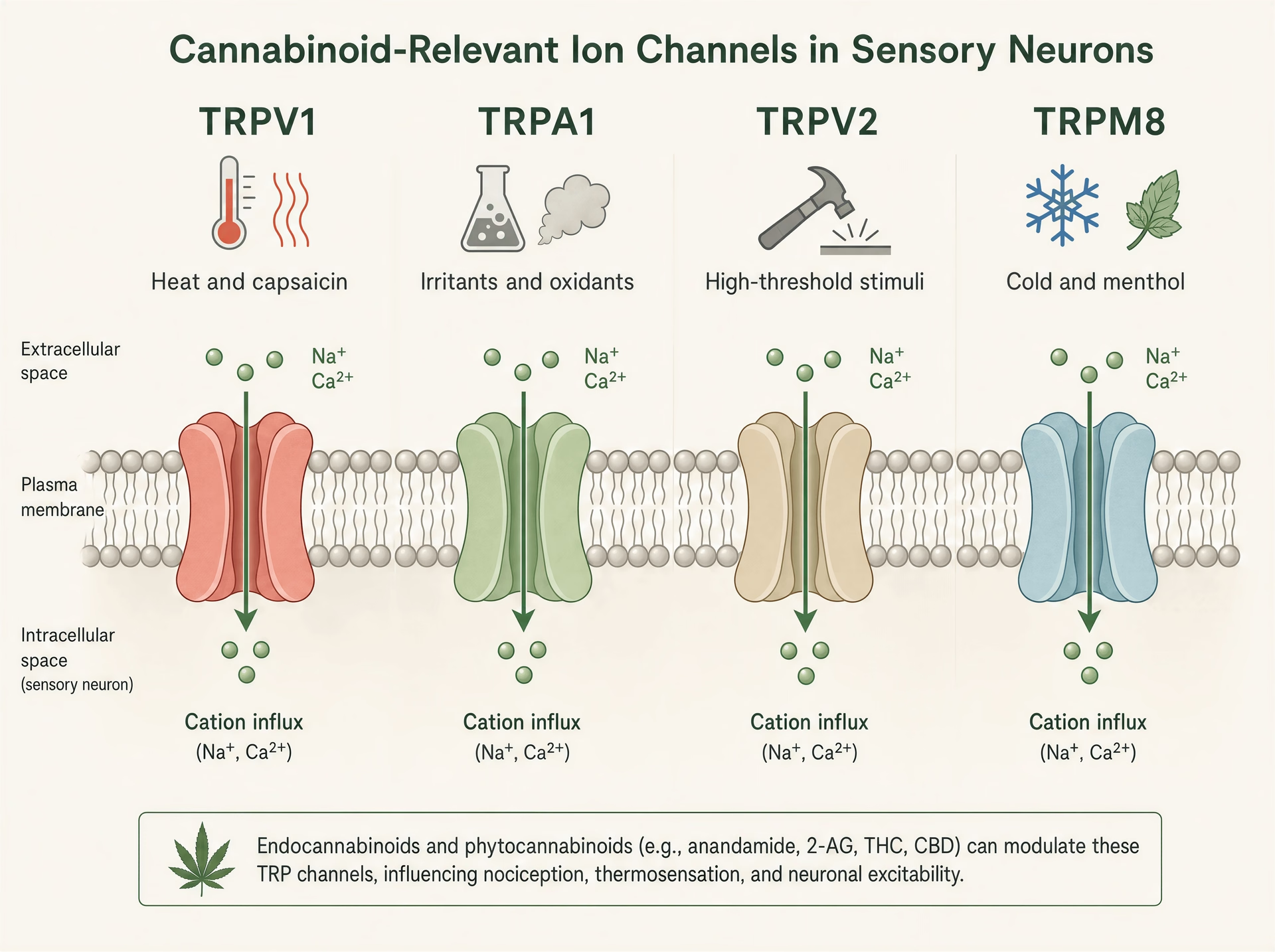
TRP-Kanäle: die Hitze-, Schmerz- und Reizsensoren, die Cannabinoide ständig treffen
Die übliche Kurzformel lautet, Cannabinoide wirkten über CB1 und CB2. Das ist zu eng, um zu erklären, was viele dieser Moleküle im Gewebe tatsächlich tun. Immer wieder treffen Phytocannabinoide auf Transiente Rezeptorpotential-Kanäle, eine Superfamilie von Ionenkanälen in Nozizeptoren, Keratinozyten, Atemwegnerven, Immunzellen und anderen sensorischen Grenzflächen, an denen der Körper Hitze, Kälte, Chemikalien, Dehnung, Verletzung und Entzündung erkennt.[7]The Nobel Prize in Physiology or Medicine 2021. The Nobel Assembly at Karolinska Institutet. Nobel Prize Press Release, 2021. https://www.nobelprize.org/prizes/medicine/2021/press-release/
Diese Biologie ist nicht obskur. Sie war in der somatosensorischen Wissenschaft so zentral, dass der Nobelpreis 2021 für Physiologie oder Medizin an David Julius und Ardem Patapoutian „for their discoveries of receptors for temperature and touch“ vergeben wurde. Julius’ Arbeit zur Identifizierung des Capsaicin-Rezeptors TRPV1 half, die moderne Sicht zu etablieren, dass Schmerzsignalgebung nicht nur ein Draht ist, der Schadensinformation transportiert; sie wird am allerersten sensorischen Ende chemisch gesteuert. Das ist für Cannabinoide wichtig, weil mehrere wichtige Pflanzen-Cannabinoide mit derselben molekularen Hardware interagieren, die auf Chili, Senföl, schädliche Hitze, Kühlmittel, saure Bedingungen und inflammatorische Lipide reagiert.
Das Ergebnis ist eine Pharmakologie, die chaotisch aussieht, wenn man einen Rezeptor und einen Effekt erwartet. Sinnvoller wird sie, wenn man in Begriffen der sensorischen Verstärkungssteuerung denkt. Viele Cannabinoide sind schwache bis mäßige Liganden an CB-Rezeptoren und zugleich direkte Modulatoren von TRP-Kanälen. Manche aktivieren sie. Manche hemmen sie. Manche tun beides je nach Konzentration, Spezies, Spleißvariante, Membranumgebung und je nachdem, ob der Assay Calcium-Einstrom, Strom, Neuropeptidfreisetzung oder Verhalten in einem Tier misst.
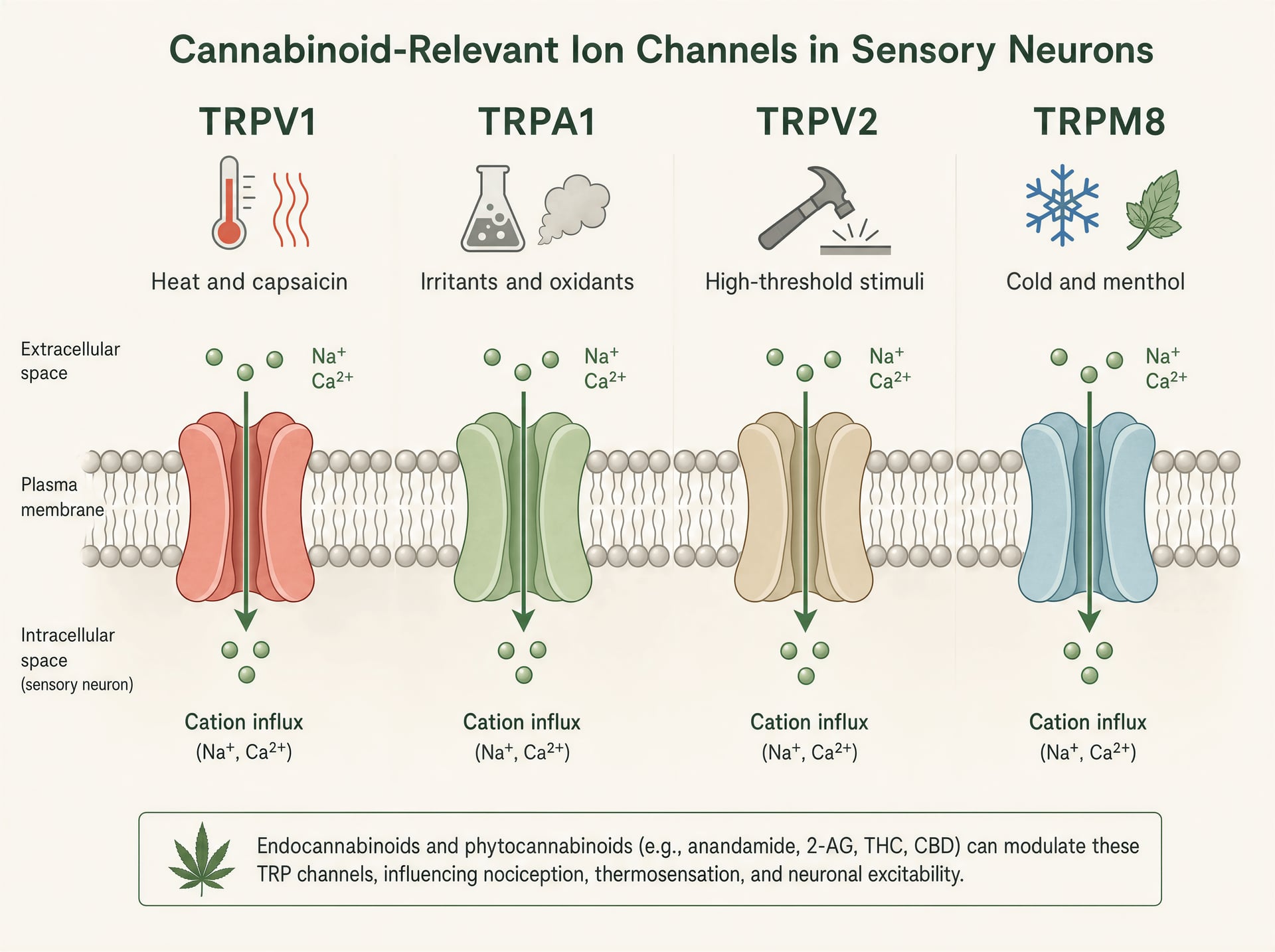
TRPV1, TRPA1, TRPV2 und TRPM8 in der sensorischen Biologie
TRP-Kanäle sind polymodale Detektoren. TRPV1 ist am besten bekannt: aktiviert durch Capsaicin, schädliche Hitze, Protonen und endogene inflammatorische Mediatoren, wird er stark in klein-durchmessrigen sensorischen Neuronen exprimiert, die Brennschmerz und neurogene Entzündung vermitteln. Öffnet man den Kanal, strömen Kationen ein, depolarisieren das Neuron und erhöhen das intrazelluläre Calcium. TRPA1 sitzt häufig in überlappenden Nozizeptor-Populationen und ist berühmt dafür, elektrophile Reizstoffe wie Allylisothiocyanat aus Senf und Wasabi, Acrolein im Rauch und oxidativen Stressprodukten, die während Entzündungen entstehen, zu detektieren. Er ist nicht nur für Schmerz relevant, sondern auch für Juckreiz, Husten, bronchiale Hyperreaktivität und migräneähnliche trigeminale Signalgebung.
TRPV2 ist weniger geradlinig. Er ist in einigen Systemen ein hochschwelliger Thermo- und Mechanosensitivitätskanal, wird aber auch in Immunzellen, Glia und proliferativen Geweben gefunden, weshalb er in Diskussionen über Entzündung und, spekulativer, Krebsbiologie immer wieder auftaucht. TRPM8 hingegen ist der kanonische Kältesensor, aktiviert durch Kälte und Verbindungen wie Menthol und Icilin. Er ist jedoch auch bei Schmerzzuständen relevant, in denen Kälteallodynie schwer werden kann, und in einigen Kontexten kann TRPM8-Aktivität Schmerz durch circuit-level Gegenstimulation dämpfen. Gleiche Familie, sehr unterschiedliche sensorische Rollen.
Diese Funktionsbreite erklärt, warum Cannabinoid-Effekte oberflächlich widersprüchlich wirken können. TRPV1 oder TRPA1 zu aktivieren kann brennen. TRPM8 zu blockieren kann Kühlungswahrnehmungen verringern, aber auch Kälteschmerz verändern. TRPV2 in einem Zelltyp zu stimulieren kann die Calcium-Signalgebung beeinflussen, ohne überhaupt einen offensichtlichen sensorischen Effekt auszulösen. Es gibt keinen einzelnen „TRP-Effekt“, genauso wenig wie es einen einzelnen „Cannabinoid-Effekt“ gibt.
CBD, CBG, CBC und THC an TRP-Familienkanälen
Unter den Phytocannabinoiden hat CBD das stärkste und am besten replizierte TRP-Profil. In heterologen Expressionssystemen aktiviert CBD humanes TRPV1, TRPA1 und TRPV2 bei mikromolaren Konzentrationen und hemmt TRPM8. Eine häufig zitierte Studie von De Petrocellis und Kollegen aus dem Jahr 2011, die Calcium-Bildgebung in transfizierten HEK-293-Zellen verwendete, fand, dass CBD als Agonist an TRPV1, TRPV2, TRPA1 und TRPV4 wirkte und zugleich TRPM8 antagonisierte. Die Potenz war nicht einheitlich: TRPA1 zeigte besonders hohe Sensitivität mit niedrig-mikromolarer Aktivität, während andere Kanäle etwas höhere Konzentrationen erforderten. Dieses Muster ist robust genug, dass TRP-Beteiligung heute Bestandteil jeder ernsthaften Darstellung der CBD-Pharmakologie ist.
CBG und CBC fügen sich in das gleiche allgemeine Muster ein, wenn auch mit eigenen Fingerabdrücken. CBG zeigte wiederholt Aktivität an TRPA1 und TRPV1 sowie eine Hemmung von TRPM8 und ist deshalb pharmakologisch interessant für Entzündungsschmerz und viszerale Hypersensitivität. CBC ist weniger gut untersucht als CBD, doch verfügbare In-vitro-Arbeiten deuten darauf hin, dass es ebenfalls TRPA1 aktiviert und TRPV1 ansprechen kann. Das sind keine kleinen Kuriositäten aus einem einzigen Assay, die nie wieder gesehen werden. Sie wiederholen sich in rekombinanten Systemen und primären sensorischen Präparationen, weshalb sie in Mechanismus-Papieren zu Analgesie und Entzündung immer wieder auftauchen.
THC ist komplizierter. Es kann TRPV2 aktivieren und wurde unter bestimmten Bedingungen auch mit TRPA1 und TRPV1 in Verbindung gebracht, doch seine Pharmakologie wird in vielen Experimenten von CB1-vermittelten Effekten dominiert, insbesondere im zentralen Nervensystem. Dennoch ist die Vorstellung falsch, THC sei nur ein CB1-Medikament. Eine 2025 von der Hebrew University berichtete Arbeit argumentierte, THC hemme periphere Nozizeptoren durch Targeting der Natriumkanäle NaV1.7 und NaV1.8, also einen separaten Nicht-CB-Mechanismus, der mit dem breiteren Punkt hier zusammenpasst: Cannabinoide treffen häufig mehrere schmerzrelevante Ziele gleichzeitig. TRP-Kanäle sind Teil dieser breiteren Nicht-CB-Karte.
Ein Vorbehalt ist nötig. Vieles dieser Evidenz stammt aus mikromolaren Assays, und nicht jeder Mikromolarwert in einer Schale entspricht einer erreichbaren freien Konzentration an einer humanen Zielstruktur. Lipophile Cannabinoide partitionieren in Membranen, binden Proteine und bilden Metaboliten; Applikationsweg und Gewebeanreicherung sind wichtig. Die Tatsache, dass eine orale CBD-Lösung von der FDA für Krampferkrankungen zugelassen ist, beweist nicht, dass TRPV1 oder TRPA1 ihre klinischen Effekte bei Epilepsie antreiben. Sie zeigt lediglich, dass CBD beim Menschen eindeutig Dinge tut, die man nicht damit erfassen kann, es als „nicht-intoxikierendes CB-Rezeptor-Molekül“ zu bezeichnen. Die molekulare Geschichte ist größer als dieses Etikett.
TRP-Aktivität ist zudem assay-sensitiv. Ein Kanal kann in einem Calcium-Assay scheinbar „aktiviert“ werden, weil intrazelluläre Speicher, Membranpotential oder endogene Lipide parallel verändert werden. Speziesunterschiede können real sein. Ebenso Zustandsabhängigkeit. Entzündetes Gewebe versauert, oxidiert und produziert Lipidmediatoren, die die TRP-Gating-Eigenschaften verändern. Ein Cannabinoid, das einen Kanal im Basalzustand kaum beeinflusst, kann an einer verletzten Nervenendigung eine viel stärkere Wirkung haben.
Desensibilisierung, Analgesie und warum Aktivierung Schmerz verringern kann
Das ist der Punkt, der Nicht-Spezialisten verwirrt: Wenn TRPV1 und TRPA1 schmerzproduzierende Kanäle sind, warum sollte ihre Aktivierung jemals Schmerz reduzieren?
Weil akute Aktivierung und anhaltende funktionelle Ausgabe nicht dasselbe sind.
TRPV1 ist das klassische Beispiel. Capsaicin brennt zunächst, desensibilisiert dann Nozizeptoren und kann nach wiederholter oder hochkonzentrierter Exposition Analgesie erzeugen. Klinisch wird dieses Prinzip beim 8%-Capsaicin-Pflaster für neuropathische Schmerzen genutzt. Der Mechanismus umfasst calciumabhängige Desensibilisierung, Erschöpfung von Neuropeptiden wie Substanz P und CGRP, veränderten Phosphorylierungszustand des Kanals und in einigen Fällen eine reversible Funktionsminderung der Nervenendigung. Ein Kanal, der anfangs stark feuert, kann später weniger reaktionsfähig werden. Das unmittelbare Signal ist pro-nozizeptiv; der nachgelagerte Zustand kann anti-nozizeptiv sein.
Cannabinoide scheinen dieselbe Logik auszunutzen. Die Aktivierung von TRPV1 oder TRPA1 durch CBD kann einen Calcium-Einstrom auslösen, gefolgt von verringerter Kanalreaktivität und gedämpfter Erregbarkeit sensorischer Neuronen. Das ist ein plausibler Weg, wie eine Verbindung in der Petrischale brennen, im Tier aber Hyperalgesie reduzieren kann. Die Zeitskala ist entscheidend. Ebenso die Dosis. Niedrige Konzentrationen können sensibilisieren oder schwach aktivieren. Höhere Konzentrationen können Desensibilisierung oder sogar breitere Membraneffekte auslösen, die das Feuern unterdrücken.
TRPA1 fügt eine weitere Ebene hinzu, weil er eng mit Entzündungsreizen und oxidativem Stress verbunden ist. In Atemwegs- und Trigeminussystemen kann wiederholte oder anhaltende Aktivierung die Neuropeptidfreisetzung und Reflexantworten verändern. Das macht ihn nicht nur für Schmerz, sondern auch für Husten, Migräne und entzündliche Schubzustände relevant. Wenn ein Cannabinoid TRPA1 aktiviert und danach die nachfolgende Reaktionsfähigkeit reduziert, kann der Nettoeffekt eine geringere Reizsignalgebung sein, auch wenn das erste molekulare Ereignis eine Kanalöffnung war.
TRPM8 zeigt in vielen Assays das umgekehrte Cannabinoid-Muster: Cannabinoide wie CBD und CBG hemmen ihn häufig statt ihn zu aktivieren. Das könnte bei Kältehypersensitivität relevant sein, in der übermäßige TRPM8-Signalgebung zu schmerzhafter Kälteallodynie beiträgt. Hier gibt es kein Paradox der Aktivierung als Linderung; die einfachere Hypothese ist eine direkte Unterdrückung eines Kältesignalwegs. Auch das sollte allerdings nicht überinterpretiert werden. In einigen Schmerzzuständen kann TRPM8-Aktivität Wärmeschmerz oder Juckreiz entgegenwirken, sodass Blockade nicht automatisch vorteilhaft ist.
Die stärkste, durch die Evidenz unterstützte Position lautet daher: TRP-Kanäle sind keine Randnotizen der Cannabinoid-Pharmakologie. Sie sind wiederkehrende, funktionell relevante Ziele, insbesondere für periphere sensorische Effekte mit Hitze, chemischer Reizung, entzündlichem Schmerz, Juckreiz und Atemwegsreflexen. Sie erklären nicht alles. Sie sind nicht immer der dominierende Mechanismus in vivo. Doch jeder, der verstehen will, warum CBD, CBG, CBC oder sogar THC Schmerz und Entzündung ohne saubere Zuordnung zu CB1 oder CB2 verändern können, braucht TRPV1, TRPA1, TRPV2 und TRPM8 früh im Bild, nicht als Nachgedanken.
Das ist auch für die Arzneimittelentwicklung wichtig. Öffentliche Gesundheitsbehörden unterscheiden bereits zwischen vertrauten Cannabinoiden und chemisch veränderten oder verstärkten Intoxikanzien, weil Zielstruktur-Unterschiede das Risiko verändern können. Dasselbe Prinzip gilt umgekehrt für Therapeutika: Wenn sich Analgesie von zentraler Intoxikation trennen lässt, besteht ein Weg darin, Verbindungen zu designen, die die Wirkung auf periphere TRP-Kanäle und andere Nicht-CB-Ziele lenken, statt stark hirngängige CB1-Agonisten zu erzeugen. Die alte, rezeptor-reduktionistische Geschichte ist für die Daten zu klein.
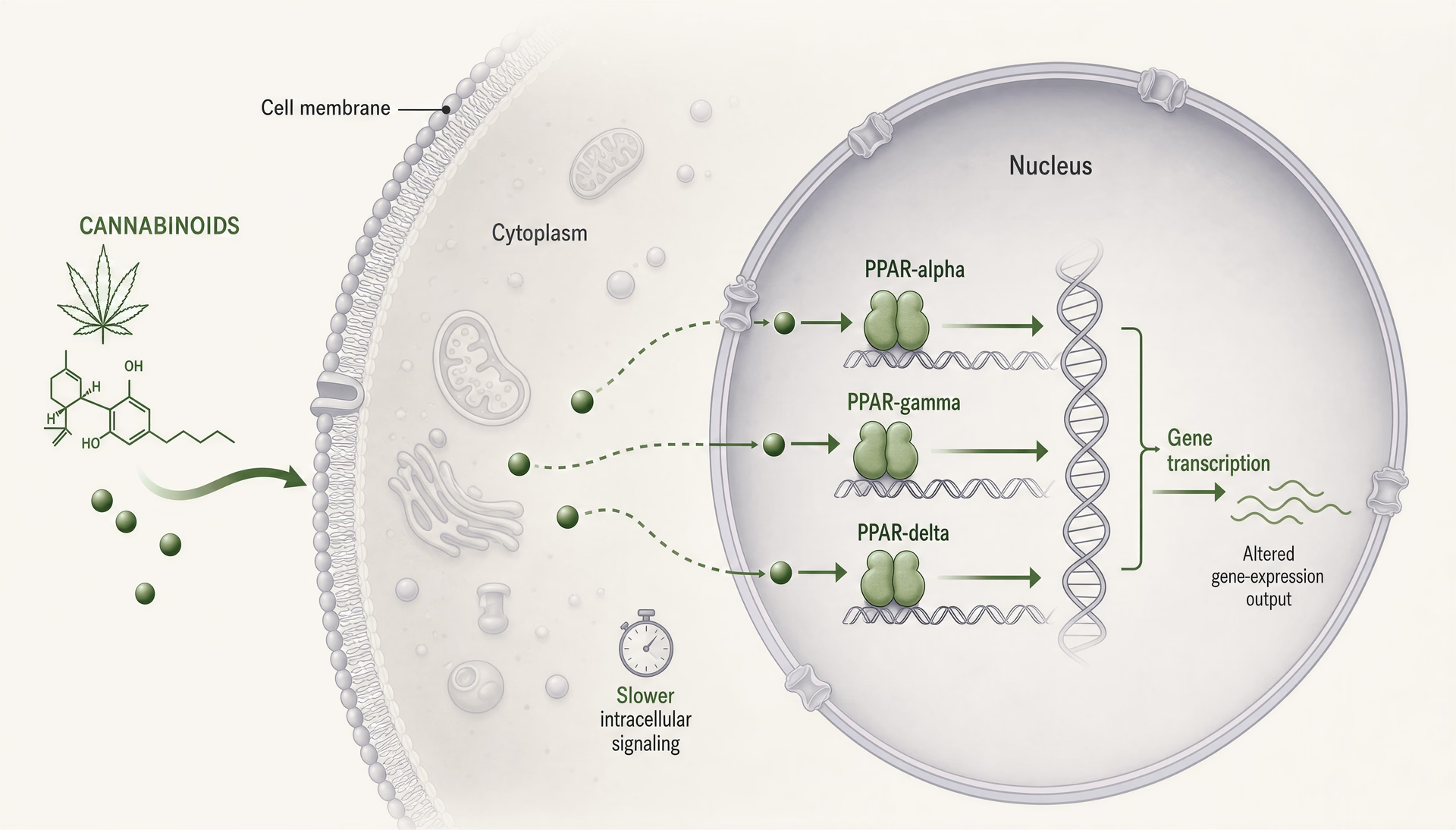
PPARs: Cannabinoide als intrazelluläre Lipidsignale, nicht nur als Liganden von Membranrezeptoren
Peroxisom-Proliferator-aktivierte Rezeptoren, meist kurz PPARs genannt, verändern die Cannabinoid-Debatte, weil sie an einem anderen Ort liegen und auf einer anderen Zeitskala arbeiten als CB1 und CB2. CB1 und CB2 sind membranständige G-Protein-gekoppelte Rezeptoren, die für schnelle Signalgebung gebaut sind: Sekunden bis Minuten, Ionenkanäle, Neurotransmitterfreisetzung, Kinasekaskaden. PPARs sind nukleäre Rezeptoren. Sie reagieren auf lipophile Moleküle, beeinflussen die Transkriptionsmaschinerie und verändern, welche Gene eine Zelle über Stunden bis Tage exprimiert. Dieser Wechsel ist wichtig. Er bedeutet, dass einige Cannabinoid-Effekte weniger wie klassische Rezeptor-Agonistik und mehr wie lipidregulierte Reprogrammierung von Entzündungstonus, mitochondrialem Handling, Fettsäureoxidation, fibrotischer Signalgebung und glialen Antworten aussehen können.
Das ist kein spekulativer Sprung. Cannabinoide sind hoch lipophil, akkumulieren in Membranen, verteilen sich in intrazelluläre Kompartimente und erzeugen Metaboliten, die andere Zielprofile haben können als das Ausgangsmolekül. Eine Wirkstoffklasse mit solchen Eigenschaften ist fast dafür gemacht, wiederholt auf nukleäre Lipidsensoren zu treffen. PPARs gehören zu den plausibelsten Orten, an denen das geschieht.
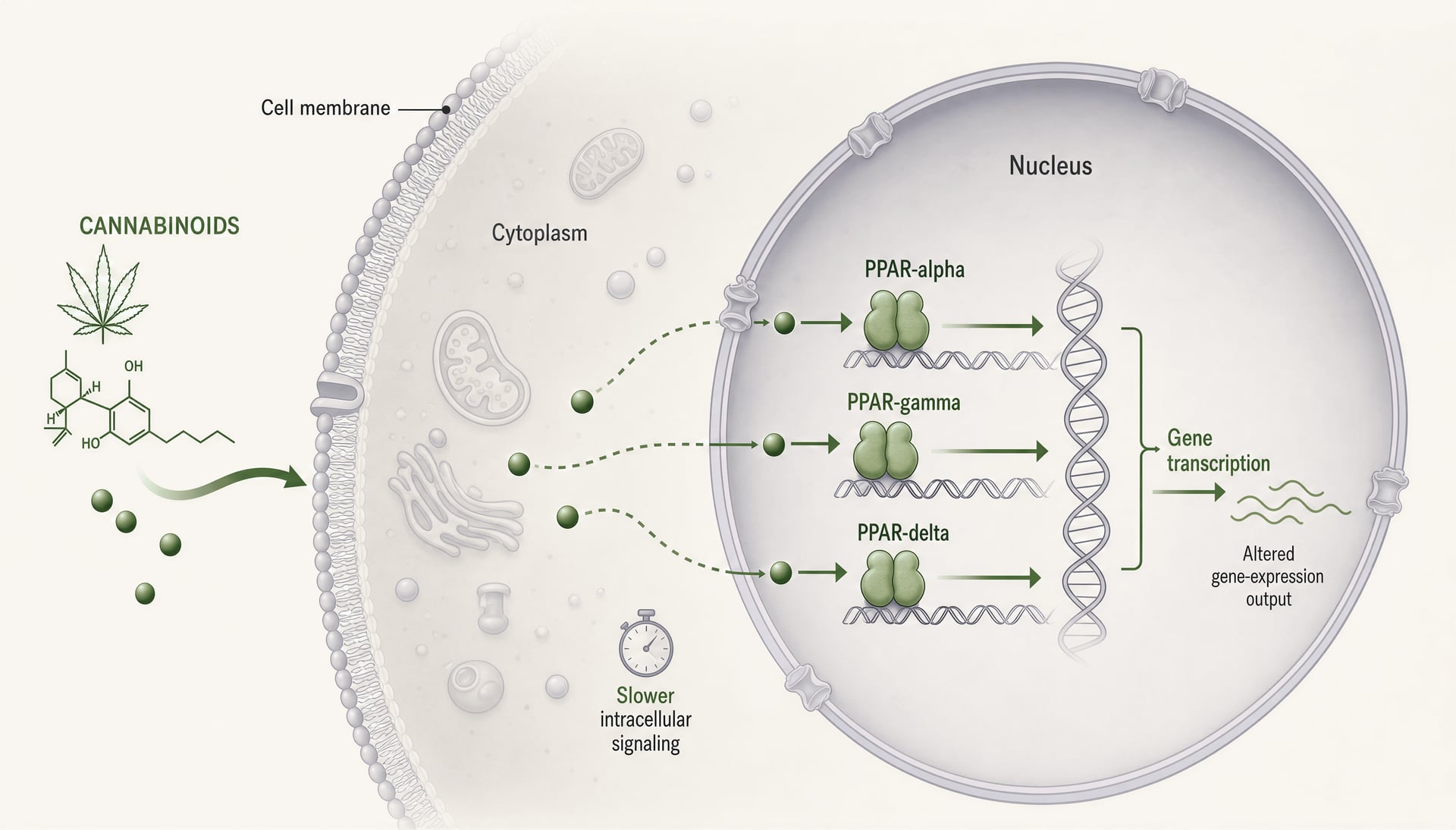
Was PPAR-alpha, PPAR-gamma und PPAR-delta tun
Die drei wichtigsten PPAR-Isoformen überschneiden sich, sind aber nicht austauschbar. PPAR-alpha ist klassisch mit Fettsäurekatabolismus verbunden. Er ist in Leber, Herz, Niere, Muskel und anderen Geweben reichlich vorhanden, die Fett intensiv verbrennen, und seine Aktivierung schiebt Transkriptionsprogramme für Beta-Oxidation, Ketogenese, Lipoproteinverarbeitung und geringere Entzündung. Pharmakologen kennen ihn von Fibrat-Medikamenten. In der Schmerz- und Entzündungsforschung ist PPAR-alpha auch außerhalb des Stoffwechsels relevant, weil er NF-kappaB-verknüpfte entzündliche Genexpression unterdrücken und sensorische Signale verändern kann.
PPAR-gamma ist die Isoform, die in Cannabinoid-Papieren immer wieder auftaucht, manchmal aus guten Gründen und manchmal, weil es die einfachste Erzählung ist. Er ist für Adipozytendifferenzierung und Insulinsensitivität hoch relevant, aber diese Kurzform unterschätzt ihn. PPAR-gamma reguliert Makrophagenpolarisation, Zytokinproduktion, oxidative Stressantworten, fibrotisches Remodeling, Endothelverhalten und Gliaaktivierung im zentralen Nervensystem. Das macht ihn offensichtlich relevant für entzündliche Darmerkrankungen, Neuroinflammation, diabetische Komplikationen und Gewebefibrose. Er ist jedoch auch ein zweischneidiges Ziel: starke Aktivierung kann die Insulinsensitivität verbessern, aber Ödeme, Gewichtszunahme und andere Risiken mit sich bringen, wie sie von Thiazolidindion-Medikamenten bekannt sind.
PPAR-delta, auch PPAR-beta/delta genannt, erhält in der öffentlichen Cannabinoid-Literatur weniger Aufmerksamkeit, sollte aber nicht übersehen werden. Er wird breit exprimiert und unterstützt Fettsäureverwertung, mitochondriale Funktion, Wundheilung, Keratinozytenbiologie und einige antiinflammatorische Programme. Je nach Kontext kann er Krankheitsprozesse bremsen oder fördern, was ein Grund dafür ist, dass die Literatur weniger ordentlich ist. Wenn ein Cannabinoid oder Cannabinoid-Metabolit PPAR-delta anspricht, kann die biologische Antwort je nach Gewebe stark variieren und sich einer einfachen „Agonist gleich Nutzen“-Geschichte entziehen.
Mechanistisch arbeiten alle drei Isoformen als ligand-aktivierte Transkriptionsfaktoren, die mit dem Retinoid-X-Rezeptor heterodimerisieren und an peroxisome proliferator response elements in der DNA binden. Wenn sie aktiviert sind, schalten sie nicht nur einen Schalter um. Sie verändern transkriptionelle Netzwerke. Co-Aktivatoren, Co-Repressoren, Chromatinzustand, Zelltyp, Entzündungskontext und ligandenspezifische Rezeptorkonformation beeinflussen das Ergebnis. Zwei Verbindungen können beide als PPAR-gamma-Agonisten bezeichnet werden und dennoch biologisch deutlich unterschiedliche Wirkungen entfalten.
Das ist für Cannabinoide besonders wichtig, weil diese oft pharmakologisch promiskuitiv sind statt saubere, einseitige Werkzeuge zu sein.
CBD und verwandte Cannabinoide in metabolischer und entzündlicher Signalgebung
CBD ist das wiederkehrende Beispiel, weil sein klinisches Profil durch CB1 oder CB2 allein schlecht erklärt wird. Die von der FDA zugelassene orale Lösung für Anfälle bei Lennox-Gastaut-Syndrom, Dravet-Syndrom und tuberösem Sklerose-Komplex zeigt, dass CBD beim Menschen pharmakologisch real ist, aber nicht, dass irgendein einzelnes Nicht-Cannabinoid-Ziel seine Wirkung erklärt. PPAR-gamma ist einer der meistgenannten Kandidaten, weil mehrere Zell- und Tierstudien CBD mit antiinflammatorischen und metabolischen Effekten verknüpft haben, die durch PPAR-gamma-Antagonisten abgeschwächt werden oder von PPAR-gamma-abhängigen transkriptionellen Veränderungen begleitet sind.
Ein häufig zitierter Beitrag von O’Sullivan und Kollegen aus dem Jahr 2009 berichtete, dass CBD eine Vasorelaxation in menschlichen Arterien auslöst und dass ein Teil dieses Effekts durch den PPAR-gamma-Antagonisten GW9662 empfindlich war, was auf eine PPAR-gamma-abhängige Komponente hindeutet. 2011 zeigten Esposito und Mitautoren in einem Alzheimer-ähnlichen Zellmodell, dass CBD die durch Beta-Amyloid induzierte Neuroinflammation reduzierte und dass die Blockade von PPAR-gamma diesen Schutzeffekt verringerte. 2013 fassten Hind und O’Sullivan Evidenz zusammen, wonach Cannabinoide PPARs direkt oder indirekt aktivieren können, und ordneten CBD, THC, Ajulemsäure, Anandamid-verwandte Lipide und mehrere synthetische Cannabinoide in diesen Rahmen ein.
Das Muster ist konsistent genug, um es ernst zu nehmen: CBD landet in experimentellen Systemen oft dort, wo Entzündungs-Gene sinken, oxidative Stressmarker abnehmen und PPAR-gamma-Antagonismus die Antwort abschwächt. Es ernst zu nehmen heißt jedoch nicht, es als gesichert zu betrachten. Viele dieser Studien verwenden mikromolare CBD-Konzentrationen. Das ist wichtig, weil intrazelluläre freie Konzentrationen in lebenden menschlichen Geweben schwer aus nominellen Bad-Konzentrationen in einer Schale abzuleiten sind. CBD bindet und stört auch Membranen, beeinflusst den Calcium-Haushalt, interagiert mit TRP-Kanälen, wirkt auf die Adenosin-Signalgebung durch Hemmung von Nukleosidtransportern und kann den Endocannabinoid-Tonus verändern. Jeder dieser Wege kann in transkriptionelle Veränderungen münden, die später „PPAR-ähnlich“ aussehen.
Verwandte Cannabinoide verstärken den Befund, bereinigen ihn aber nicht. THC wurde in einigen Systemen als PPAR-gamma-aktivierend beschrieben, allerdings meist schwach im Vergleich zu spezialisierten Liganden. Cannabidiolsäure und Tetrahydrocannabinolsäure zeigten in ausgewählten Assays PPAR-Aktivität. Endocannabinoid-verwandte Lipide wie Palmitoylethanolamid, Oleoylethanolamid und einige oxidierte Derivate weisen stärkere und etabliertere Beziehungen zu PPAR-alpha und PPAR-gamma auf als die bekannteren Phytocannabinoide. Das ist ein Grund, warum der Rahmen der intrazellulären Lipidsignalgebung besser ist als ein enger Rahmen „Pflanzen-Cannabinoide binden PPARs“. Die aktive Spezies kann das Ausgangscannabinoid, ein Metabolit, ein ko-verabreichtes Lipidmediator-Molekül oder eine nachgeschaltete Veränderung endogener Lipidpools sein.
Ajulemsäure ist eine nützliche Fallstudie. Sie ist ein synthetisches Analogon mit THC-Bezug, wurde aber bewusst weg von klassischer Intoxikation entwickelt. In präklinischen Arbeiten zeigte sie antiinflammatorische und antifibrotische Wirkungen, mit Evidenz, die unter anderem PPAR-gamma impliziert. Diese Art von Medizinalchemie spiegelt einen breiteren Trend im Feld wider. Bereits 2016 reflektierte ein ACS-Journal-of-Medicinal-Chemistry-Paper mit dem Titel „Library Docking for Cannabinoid-2 Receptor Ligands“ eher struktur-basierte Zielentwicklung als grobe Rezeptorbezeichnungen, und neuere Cannabinoid-Programme zielen zunehmend darauf ab, Analgesie, Anxiolyse oder Immunmodulation von zentraler CB1-Aktivierung zu trennen. Dieselbe Logik gilt für PPAR-aktive Gerüste: Wenn nützliche Cannabinoid-Biologie über transkriptionelle und periphere Mechanismen extrahiert werden kann, gibt es keinen Grund, warum Arzneimittelentwicklung in THC-ähnlicher Pharmakologie gefangen bleiben sollte.
Die Daten zu CBD in der metabolischen Signalgebung sind gemischter als die zu seinen antiinflammatorischen Effekten. Einige präklinische Studien deuten auf verbesserte Insulinsensitivität, reduzierte entzündliche Adipokine oder besseres mitochondriales Handling hin. Andere zeigen keinen wesentlichen Nutzen, und die humanen Daten sind dünn. Die öffentliche Diskussion eilt hier oft den Daten voraus. Die Tatsache, dass PPAR-gamma Glukose- und Fettgewebebiologie kontrolliert, bedeutet nicht, dass CBD beim Menschen in Standardexpositionen ein klinisch bedeutsamer Stoffwechselmodulator ist.
Gen-Transkription, verzögerte Effekte und Grenzen der Evidenz
PPAR-Biologie erzwingt eine Korrektur der Zeitachse. Wenn ein Cannabinoid-Effekt innerhalb von Sekunden oder wenigen Minuten auftritt, sind PPARs als Primärerklärung unwahrscheinlich. Nukleäre Rezeptorsignalgebung erfordert im Allgemeinen Zugang des Liganden zu intrazellulären Kompartimenten, Rezeptorbindung, veränderte Rekrutierung von Co-Regulatoren, transkriptionelle Veränderungen und dann Folgen auf Proteinebene. Das braucht Zeit. Stunden sind plausibel. Tage sind häufig. Wenn Arbeiten behaupten, eine schnelle Cannabinoidwirkung laufe „über PPAR-gamma“, ist Skepsis angebracht, sofern das Design nicht klar unmittelbare nicht-genomische Signalgebung von späteren transkriptionsabhängigen Effekten trennt.
Das Assay-Design ist das wiederkehrende Problem. Reporter-Assays können zeigen, dass eine Verbindung die PPAR-abhängige Transkription steigert, aber Reporter-Systeme sind künstlich und können schwache Aktivität überzeichnen. Antagonistenstudien sind informativ, doch Medikamente wie GW9662 sind kein magisches Wahrheitsserum; Off-Target-Effekte und partielle Blockade verkomplizieren die Interpretation. Bindungsassays helfen, aber direkte Bindung garantiert nicht, dass die Gewebeexposition die nötige Konzentration in vivo erreicht. Knockout-Modelle sind stärker, doch Kompensation durch andere Wege kann die Ergebnisse verwischen. Die beste Evidenz kombiniert Methoden: direkte Zielstruktur-Interaktion, rezeptorselektive Pharmakologie, genetische Störung, relevante Gewebekonzentrationen und eine zur Transkriptionswirkung passende Zeitachse. Vieles der Cannabinoid-PPAR-Literatur erreicht diesen Standard nicht.
Die Prominenz von PPAR-gamma in der CBD-Forschung ist daher sowohl gerechtfertigt als auch übertrieben. Gerechtfertigt, weil das Signal in vaskulären, entzündlichen, neurodegenerativen und fibrosebezogenen Modellen wiederkehrt. Übertrieben, weil CBD genau die Art von lipophilem, multi-target Molekül ist, bei dem intrazelluläre Konzentration, aktive Metaboliten und Assay-Kontext verführerische, aber unvollständige mechanistische Geschichten erzeugen können. Ein Abfall von TNF-alpha oder IL-6 nach CBD-Exposition ist kein Fingerabdruck. Es ist ein Hinweis.
Trotzdem bleibt der größere Punkt bestehen. Cannabinoide sollten nicht nur als Liganden von membranständigen Cannabinoid-Rezeptoren behandelt werden. Einige wirken direkt oder indirekt als intrazelluläre Lipidsignale, die nukleäre Transkriptionsmaschinerie aktivieren können. Das eröffnet plausible Wege zu antiinflammatorischen, antifibrotischen und neuroimmunen Effekten, die langsamer, weniger an Intoxikation gebunden und potenziell relevanter für langfristige Krankheitsmodifikation sind als die akute CB1-Signalgebung. Es wirft auch eine regulatorische Lehre auf. Wie Behörden bereits in anderen Kontexten betont haben, einschließlich der HHS-Erklärung von 2025, dass verstärkte 7-Hydroxymitragynin-Produkte „an imminent hazard to public safety“ darstellen, sind molekulare Unterschiede entscheidend. Kleine Strukturänderungen können die Zielbindung umlenken. Für Cannabinoide und cannabinoidähnliche Produkte bedeutet das, dass Sicherheits- und Wirksamkeitsfragen nicht aus THC-Vertrautheit abgeleitet werden können, und die PPAR-Biologie ist einer der Gründe.
GPR55, GPR18 und GPR119 und das Orphan-GPCR-Problem
Ein Orphan-GPCR ist ein G-Protein-gekoppelter Rezeptor, dessen endogener Ligand, physiologische Rolle oder beides unklar bleibt. Ein deorphanisierter Rezeptor ist einer, für den ein überzeugender endogener Aktivator vorgeschlagen und so gut repliziert wurde, dass eine Arbeitsbiologie tragfähig ist. Das klingt sauber. In der Praxis ist es das selten. Die Cannabinoid-Pharmakologie läuft immer wieder in dieses Problem hinein, weil Endocannabinoide und Phytocannabinoide lipophil, membranaktiv und promiskuitiv sind: Sie können Calciumfluss, Kinaseaktivität oder Transkription auf eine Weise verändern, die rezeptorvermittelt aussieht, selbst wenn das direkte Ziel ungesichert ist. Genau so gelangten GPR55, GPR18 und GPR119 als „nichtklassische Cannabinoid-Rezeptoren“ in die Diskussion.
Die Versuchung, ein neues Rezeptor-Label zu prägen, ist groß. Es macht Schlagzeilen. Es geht jedoch schneller als die Evidenz. GPR55 kam dem Etikett „CB3“ am nächsten, doch das Feld erreichte nie die Kohärenz, die CB1 und CB2 stützte. Dieselbe Vorsicht gilt noch stärker für GPR18 und GPR119.
Warum GPR55 einst als möglicher Cannabinoid-Rezeptor galt
GPR55 wurde 1999 kloniert, und frühe Expressionsanalysen lokalisierten ihn in Geweben, die für die Cannabinoid-Biologie relevant sind: Gehirnregionen, dorsale Wurzelganglien, Milz, Gastrointestinaltrakt, Gefäße, Immunzellen und knochenbezogene Zellen einschließlich Osteoklasten und osteoblastärer Vorläuferpopulationen. Diese Verteilung war bedeutsam. Ein Rezeptor, der in Schmerzbahnen, entzündlichem Gewebe und Knochen exprimiert wird, lädt sofort zum Vergleich mit CB1 und CB2 ein, insbesondere wenn Cannabinoid-Liganden seine Messwerte zu verschieben scheinen.
Auch sein Signalprofil wirkte anders genug, um interessant zu sein. Anders als CB1 und CB2, die hauptsächlich Gi/o koppeln und tendenziell die Adenylylcyclase hemmen, signalisiert GPR55 meist über Gα12/13 und manchmal über Gq-verknüpfte Wege und aktiviert RhoA, Phospholipase C, ERK und intrazelluläre Calciumfreisetzung. In Zellassays ist das charakteristische Messsignal oft ein Calcium-Transient. Das machte GPR55 in heterologen Systemen leicht „sichtbar“, aber auch leicht überzuinterpretieren, weil Calcium-Assays empfindlich auf Rezeptordichte, Zellhintergrund, Ligandenlipophilie und Assayzeitpunkt reagieren.
Der konkrete Grund, warum GPR55 ein Cannabinoid-Rezeptorkandidat wurde, lag darin, dass mehrere Cannabinoide und cannabinoidähnliche Liganden messbare Effekte daran zeigten. Ryberg und Kollegen berichteten 2007 im British Journal of Pharmacology, dass GPR55 durch mehrere Cannabinoid-Liganden aktiviert werden könne, und schlugen ihn als „a novel cannabinoid receptor“ vor. Dieses Paper wurde zum historischen Dreh- und Angelpunkt. Es entschied die Frage nicht; es erzeugte sie.
Bald danach zeigten sich die Brüche. Einige Gruppen fanden, dass Lysophosphatidylinositol, insbesondere 2-Arachidonoyl-LPI-Spezies, ein überzeugenderer endogener Agonist war als jedes klassische Cannabinoid. Oka und Kollegen 2007 und spätere Arbeiten vertraten diese Ansicht nachdrücklich. Andere beobachteten, dass in der Cannabinoidforschung häufig diskutierte Verbindungen an GPR55 inkonsistent wirkten: Cannabidiol (CBD) erschien in einigen Assays häufig als Antagonist oder negativer Modulator, während Delta-9-THC je nach System schwach, partiell oder inaktiv war. Abnormes Cannabidiol, O-1602 und bestimmte synthetische Cannabinoide zeigten teils klarere Aktivität als THC selbst. Das ist nicht das, was man von einem sauberen dritten Cannabinoid-Rezeptor erwarten würde.
Dennoch ist GPR55-Biologie real, auch wenn das Label instabil ist. In der Schmerzforschung wird der Rezeptor in sensorischen Neuronen und spinalen Schaltkreisen exprimiert, und genetische oder pharmakologische Unterbrechung der GPR55-Signalgebung verringerte in einigen Nagermodellen die mechanische Überempfindlichkeit. Staton und Kollegen verknüpften in Pain (2008) GPR55-Aktivierung mit entzündlicher und neuropathischer Schmerzverarbeitung, wobei Antagonismus die Hypersensitivität reduzierte. Doch der Effekt ist nicht in allen Modellen oder mit allen Liganden universell. Einige Daten deuten auf pronozizeptive Signalgebung über Calcium-Mobilisierung und erhöhte neuronale Erregbarkeit hin; andere Datensätze sind schwächer oder modellbegrenzt. Die sicherste Lesart lautet, dass GPR55 in manchen Kontexten zur Schmerzsignalgebung beitragen kann, insbesondere in Entzündungszuständen, aber kein Meister-Schmerzschild ist.
Die Knochenbiologie liefert ein robusteres Signal. Warum? Weil GPR55-Knockout-Phänotypen schwerer als Assay-Artefakte abzutun sind. 2009 berichteten Whyte und Kollegen in PNAS, dass Mäuse ohne GPR55 eine erhöhte Knochenmasse und eine beeinträchtigte Osteoklastenfunktion aufwiesen, was dafür spricht, dass GPR55 die Osteoklastenresorption fördert. Das passte mechanistisch zu seiner Calcium- und RhoA-gekoppelten Signalgebung. Osteoklasten sind auf Zytoskelett-Umlagerungen und lokales Calcium-Handling angewiesen; GPR55 passt besser zu dieser Maschinerie als CB1. Wenn ein Cannabinoid oder cannabinoidähnliches Molekül hier GPR55 moduliert, kann die physiologische Folge erheblich sein.
Entzündung ist das dritte große Thema. GPR55 ist in immunrelevanten Zellen vorhanden, und seine Aktivierung wurde mit Zytokinfreisetzung, Leukozytenverhalten und vaskulären Entzündungsreaktionen verknüpft. Doch auch hier ist die Richtung nicht vollkommen einheitlich. In manchen Präparationen wirkt GPR55-Aktivierung proinflammatorisch, in anderen eher regulierend, was vermutlich Zelltyp, Liganden-Bias und Rezeptor-Crosstalk widerspiegelt und nicht bloße Widersprüchlichkeit. Ein Rezeptor, der über mehrere Wege koppelt und in unterschiedlichen Membranumgebungen sitzt, erzeugt kein universelles Ergebnis.
Diese Komplexität erklärt den langjährigen Agonist/Antagonist-Streit in der Cannabinoid-Literatur. CBD ist das klarste Beispiel. Über mehrere Studien hinweg verhielt sich CBD oft als GPR55-Antagonist oder funktioneller Inhibitor und dämpfte LPI-getriebene Calcium-Signalgebung. Lauckner et al. zeigten 2008 in einem weithin zitierten PNAS-Paper, dass GPR55-Aktivierung den intrazellulären Calciumspiegel erhöhte und Neurotransmitterfreisetzung förderte, während CBD Aspekte dieser Signalgebung entgegengesetzt beeinflusste. Daraus entstand die anhaltende Hypothese, dass einige CBD-Effekte, insbesondere in Anfalls- und Entzündungsmodellen, teilweise auf GPR55-Blockade statt auf CB1- oder CB2-Wirkung beruhen könnten. Diese Idee ist plausibel. Sie ist beim Menschen nicht als dominanter Mechanismus bewiesen.
THC ist noch unübersichtlicher. Einige Berichte klassifizieren es als GPR55-Agonisten mit niedriger Potenz; andere finden nahezu keine Wirksamkeit; wieder andere vermuten ein Verhalten, das von Rezeptorreserve oder gemessener Signalbahn abhängt. Ein Ligand kann in einem β-Arrestin-Assay als Agonist, in der Bindung neutral und in einem Calcium-Assay antagonistisch erscheinen, wenn das System überexprimiert oder verzerrt ist. Das ist keine technische Fußnote. Das ist die Geschichte.
Die gemischte Evidenz für GPR18 und GPR119
GPR18 wurde oft diskutiert, weil er in einigen Systemen auf N-Arachidonoyl-Glycin anspricht, ein endocannabinoidbezogenes Lipid, und weil abnormes Cannabidiol und verwandte Verbindungen vaskuläre oder immunologische Effekte zeigten, die manche Autoren GPR18 zuordneten. Die Expression wurde in Immunzellen, Mikroglia, Milz und einigen peripheren Geweben beschrieben. Das machte ihn attraktiv als Kandidat für Entzündungsregulation, Immunzellwanderung und möglicherweise Schmerz.
Doch die Pharmakologie war von Anfang an uneinheitlich. Kohno und Kollegen stützten 2006 die GPR18-Aktivierung durch N-Arachidonoyl-Glycin. McHugh und Kollegen verknüpften GPR18 später mit Mikroglia-Migration und Entzündungssignalgebung. Dann traten Replikationsprobleme auf. Einige Labore konnten die Ligandenreaktionen in transfizierten Systemen nicht reproduzieren. Andere fanden starke Abhängigkeit von Rezeptortagging, Zelllinie oder Speziesortholog. Ein Rezeptor, der nur in einer Assay-Architektur „funktioniert“, ist in keinem stabilen Sinne deorphanisiert. Für Cannabinoide ist die Evidenz schwächer, als populäre Zusammenfassungen suggerieren. Es kann echte Biologie geben, aber der Fall für GPR18 als bona fide Cannabinoid-Rezeptor bleibt dünn.
GPR119 ist anders. Er ist als Cannabinoid-Rezeptor deutlich weniger plausibel, trotz gelegentlicher Aufnahme in breite „Nicht-CB“-Rezeptorlisten. GPR119 ist vor allem mit Lipidsensing in pankreatischen Beta-Zellen und enteroendokrinen Zellen assoziiert, koppelt über Gs an einen erhöhten cAMP-Spiegel und fördert glukoseabhängige Insulinsekretion und Inkretinfreisetzung. Oleoylethanolamid ist ein besser etablierter endogener Liganden-Kandidat als irgendein klassisches Cannabinoid. Da einige Fettsäureethanolamide strukturell der Endocannabinoid-Chemie nahe stehen, kann GPR119 aus Assoziation in Cannabinoid-Diskussionen hineingezogen werden. Das ist größtenteils Kategorienverwechslung. Die Überlappung ist chemische Nachbarschaft, kein starker Beleg dafür, dass THC, CBD oder wichtige Phytocannabinoide in physiologischen Konzentrationen wesentlich über GPR119 wirken.
Was Orphan-Rezeptor-Pharmakologie in Schlagzeilen falsch macht
Das Standardversagen der Medien ist simpel: Ein positiver Signalassay wird zu „Wissenschaftler entdeckten einen neuen Cannabinoid-Rezeptor“. Dieser Sprung ignoriert mindestens vier Filter.
Erstens die Assay-Abhängigkeit. Calcium-Mobilisierung, β-Arrestin-Rekrutierung, ERK-Phosphorylierung, dynamische Massenumverteilung und Radioligandenbindung stellen nicht dieselbe Frage. Ein lipophiler Ligand kann Membranen stören, den Rezeptor-Transport verändern oder Pathway-Bias zeigen. Wird der Rezeptor überexprimiert, erscheinen schwache Verbindungen plötzlich stark.
Zweitens Speziesunterschiede. Humanes GPR55 ist pharmakologisch nicht in jedem Detail identisch mit murinem GPR55, und Gleiches gilt für GPR18. Ein Ligandenprofil, das in HEK293-Zellen mit dem humanen Rezeptor erstellt wurde, muss keine Ratten-Schmerzstudie vorhersagen.
Drittens die Konzentration. Viele Cannabinoid-Arbeiten berichten in vitro mikromolare Aktivität. Das kann pharmakologisch relevant sein, aber nicht automatisch. Gewebespiegel nach Inhalation, oraler Gabe, First-Pass-Metabolismus oder lokaler Fettanreicherung variieren enorm. In-vitro-Bindung ist kein klinischer Mechanismus.
Viertens der Kontext. Ein Rezeptor in Immunzellen kann einen Effekt vermitteln; derselbe Rezeptor in Osteoklasten einen anderen. Ergänzt man das um Crosstalk mit TRP-Kanälen, PPARs, Serotoninrezeptoren und sogar Natriumkanälen, bricht die schöne Ein-Ligand-ein-Rezeptor-Geschichte schnell zusammen.
Deshalb hat sich „CB3“ nie durchgesetzt. GPR55 hat glaubwürdige Biologie in Calcium-Signalgebung, Schmerz, Knochenumbau und Entzündung. Er hat jedoch auch widersprüchliche Cannabinoid-Pharmakologie, starke Assay-Sensitivität und einen starken Konkurrenzanspruch, dass LPI-Familienlipide seine primären physiologischen Liganden sind. GPR18 ist noch unsicherer. GPR119 gehört größtenteils nicht in denselben Korb, außer als Erinnerung daran, dass lipidbasierte GPCRs leicht mit Cannabinoiden assoziiert werden.
Für die Cannabinoid-Wissenschaft lautet die Lehre: Zurückhaltung. Diese Rezeptoren können sehr wichtig sein. Sie rechtfertigen jedoch keine voreilige Umbenennung.
Serotonin-Signalgebung: wo Cannabinoide mit 5-HT-Systemen zusammentreffen
Serotonin ist der Punkt, an dem viele populäre CBD-Behauptungen gleichzeitig plausibler und glitschiger werden. Der plausible Teil ist einfach: In Zellassays, Ratten-Angstmodellen, Stressparadigmen und einer kleinen Zahl menschlicher experimenteller Studien taucht 5-HT1A immer wieder als bedeutsamer Knotenpunkt der Verhaltenseffekte von CBD auf. Der glitschige Teil besteht darin, dass „wirkt auf Serotonin“ mehrere verschiedene Dinge bedeuten kann. Es könnte direkte Agonistik an der orthosterischen Stelle bedeuten. Es könnte positive allosterische Modulation bedeuten. Es könnte eine Förderung der Rezeptorsignalgebung ohne hochaffine Bindung bedeuten. Oder es könnte bedeuten, dass CBD die Netzwerkaktivität vor- oder nachgeschalteter serotonerger Neurone verändert und dadurch ein serotoninabhängiges Ergebnis erzeugt, ohne überhaupt ein klassisches Serotoninrezeptor-Medikament zu sein.
Diese Unterscheidung ist enorm wichtig. Wenn eine Verbindung das Verhalten auf eine Weise beruhigt, die durch einen 5-HT1A-Antagonisten wie WAY-100635 blockiert wird, beweist das allein noch nicht, dass die Verbindung ein 5-HT1A-Agonist ist. Es beweist die Abhängigkeit von 5-HT1A-Signalgebung in diesem Modell. Das ist nicht dieselbe Behauptung, und die Cannabinoid-Berichterstattung vermischt diese Aussagen oft.
5-HT1A und die Angstfrage
Der stärkste Serotoninbezug für Cannabinoide, insbesondere CBD, ist 5-HT1A. Dieser Rezeptor ist ein Gi/o-gekoppelter Serotoninrezeptor, der sowohl als Autorezeptor auf raphe-serotonergen Neuronen als auch als postsynaptischer Rezeptor in angstrelevanten Regionen wie Hippocampus, Amygdala und präfrontalem Kortex exprimiert wird. Arzneimittel, die dieses System aktivieren oder rekrutieren, können in manchen Situationen Angst reduzieren, aber die Rezeptorlage ist wichtig: Die serotonerge Feuerung über Autorezeptoren herunterzufahren ist nicht dasselbe wie postsynaptische Signalgebung in limbischen Schaltkreisen zu gestalten.
CBD gelangte über präklinische Arbeiten in den 2000er- und 2010er-Jahren in diese Diskussion, die anxiolytikaähnliche Effekte in Tests wie Elevated Plus Maze, Vogel-Konflikt-Test und kontextuellen Furchtparadigmen zeigten, mit teilweiser Blockade durch WAY-100635. Ein häufig zitierter Beitrag ist Campos und Guimarães, 2008, der fand, dass intra-prelimbisches CBD stressbedingte kardiovaskuläre Reaktionen reduzierte und dass 5-HT1A-Mechanismen zu diesem Effekt beitrugen. Eine weitere wichtige Humanstudie ist Bergamaschi et al., 2011: In einem simulierten Vortrag vor Publikum reduzierte 600 mg orales CBD die Angst bei Probanden mit sozialer Angststörung im Vergleich zu Placebo. Dieses Paper bewies keine 5-HT1A-Vermittlung beim Menschen, passte jedoch zum präklinischen Muster und machte Serotonin zu einem ernstzunehmenden Mechanismus-Kandidaten statt zu einer Marketingformel.
Die Rezeptorpharmakologie mündete jedoch nie in eine einfache Erzählung „CBD ist ein Serotonin-Agonist“. Frühe In-vitro-Arbeiten legten nahe, dass CBD an menschlichen 5-HT1A-Rezeptoren Liganden verdrängen und in einigen Signalassays als Agonist wirken könne, doch die Affinitäten waren mäßig und assayabhängig. Russo und Kollegen berichteten 2005, dass CBD in [35S]GTPγS-Bindungsassays als Agonist an klonierten humanen 5-HT1A-Rezeptoren wirke. Dieser Befund war einflussreich, aber spätere Arbeiten machten ihn komplizierter. Einige Gruppen sahen schwache direkte Aktivität. Andere sahen funktionelle Verstärkung, die besser durch allosterische oder Membran-Effekte erklärt wurde. Die Literatur ist nur in einem Punkt konsistent: 5-HT1A ist für die angstbezogene Pharmakologie von CBD wichtiger, als CB1 oder CB2 allein erklären können.
Deshalb scheitert der Rezeptor-Reduktionismus. Wäre CBD einfach ein sauberer 5-HT1A-Agonist, müsste sein Profil bekannten serotonergen Anxiolytika viel ähnlicher sein als tatsächlich. Stattdessen ist das Verhaltenssignal stark kontextabhängig und zeigt oft invertierte U-förmige Dosis-Wirkungs-Kurven. In einigen Nagertests reduzieren moderate Dosen angstähnliches Verhalten, während niedrigere oder höhere Dosen weniger bewirken. Das ist ein Warnsignal gegen Ein-Rezeptor-Erzählungen. Eine mögliche Ursache ist die TRPV1-Aktivierung bei höheren Konzentrationen. Ebenso Effekte auf Endocannabinoid-Tonus, Adenosin-Aufnahme und intrazelluläres Calcium. Ein Molekül kann 5-HT1A rekrutieren und sich trotzdem weigern, wie ein Lehrbuch-5-HT1A-Medikament zu wirken.
Direkte Bindung versus indirekte serotonerge Effekte
Die beste Lesart der Serotonin-Evidenz ist stufenweise. Auf molekularer Ebene gibt es Unterstützung für eine direkte Interaktion zwischen CBD und 5-HT1A, aber nicht für die Art saubere, hochaffine, hochwirksame Interaktion, die die Frage abschließend beantwortet. Je nach Assaysystem wurde CBD als schwacher Agonist, partieller Agonist oder positiver allosterischer Modulator beschrieben. Die Uneinigkeit ist nicht bloße Semantik. Orthosterische Agonisten besetzen die Hauptbindungsstelle des Serotonins. Positive allosterische Modulatoren verändern das Rezeptorverhalten von einer anderen Stelle und können die endogene Serotoninantwort verstärken, ohne den Rezeptor selbst stark zu aktivieren. Diese Mechanismen haben unterschiedliche Konsequenzen für Dosis, Zeitverlauf, Nebenwirkungen und die Übertragung auf den Menschen.
Signalübertragungsdaten sprechen oft eher für eine Förderung als für brute-force Aktivierung. In einigen Präparationen verstärkt CBD 5-HT1A-vermittelte Kaskaden, einschließlich Effekten auf ERK und andere nachgeschaltete Wege, stärker als es seine schwache Bindung allein erwarten ließe. Dafür gibt es mehrere mögliche Erklärungen. CBD ist stark lipophil und partitioniert in Membranen, wo es das Rezeptorumfeld und die G-Protein-Kopplung verändern kann. Es kann auch Anandamid-Signalgebung indirekt erhöhen, und der Crosstalk zwischen Endocannabinoid- und Serotoninsystem im dorsalen Raphe und Vorderhirn ist gut dokumentiert. Dann ist da Adenosin: CBD hemmt in einigen Systemen die Aktivität von Nukleosidtransportern und erhöht dadurch extrazelluläres Adenosin, was die neuronale Erregbarkeit in einer Weise verändert, die in serotonerge Schaltkreise hineinwirken kann. Nichts davon macht 5-HT1A irrelevant. Es zeigt vielmehr, dass der Rezeptor eingebettet ist.
Tierpharmakologie liefert stärkere Hinweise auf Serotonin-Abhängigkeit als auf direkte Agonistik. Immer wieder schwächt WAY-100635 die Wirkungen von CBD in Angst-, Panik-, Übelkeits- und Stressmodellen ab. Resstel et al., 2009, verknüpften etwa die Abschwächung akuter Stressreaktionen durch CBD mit 5-HT1A-Mechanismen. Auch die Arbeiten von Rock und Parker zu Übelkeit und antizipatorischer Übelkeit bei Nagetieren implizierten 5-HT1A im antiemetischen Profil von CBD. Das sind nützliche Ergebnisse, sollten jedoch als Wegweiser gelesen werden. Wenn die Blockade von 5-HT1A den Effekt beseitigt, ist der Weg beteiligt. Sie legt jedoch nicht fest, ob der Rezeptor direkt gebunden, allosterisch moduliert oder über Schaltkreisveränderungen rekrutiert wird.
Die menschliche Evidenz bleibt bescheiden. Die Bergamaschi-Studie von 2011 wird oft zitiert, weil sie ein messbares anxiolytisches Signal bei sozialer Angst während des Vortrags vor Publikum zeigte. Kleinere Bildgebungsstudien berichteten, dass CBD limbische und paralimbische Aktivierung während emotionaler Verarbeitung verändert. Keine dieser Studien identifizierte jedoch in Menschen eine 5-HT1A-Rezeptorbelegung, wie PET-Studien es für etablierte serotonerge Medikamente leisten können. Dieses Fehlen ist wichtig. Wir leiten Mechanismen aus Übereinstimmungen ab, wir messen sie nicht direkt bei klinischen Dosen.
Die beruhigenden Effekte von CBD hängen teilweise von der 5-HT1A-Signalisierung ab.Limited evidence
Warum das beruhigende Profil von CBD sich einfachen Rezeptorlabels entzieht
CBD hat bereits eine FDA-zugelassene Anwendung, und das ist nicht Angst. Die FDA-Kennzeichnung von 2024 für Cannabidiol-Orallösung beschränkt die Indikation auf Anfälle im Zusammenhang mit dem Lennox-Gastaut-Syndrom, dem Dravet-Syndrom oder dem tuberösen Sklerose-Komplex bei Patienten ab 1 Jahr. Diese Tatsache ist eine nützliche Korrektur gegen Übertreibung. Eine Verbindung kann glaubwürdige anxiolytische Signale haben, ohne dass die Wirksamkeit gegen Angst regulatorisch abgesichert ist, und sie kann serotonerg beteiligt sein, ohne sauber in die Schublade „Serotonin-Medikament“ zu passen.
Ein Teil des Problems ist die Skala. In vitro sind Cannabinoide pharmakologisch unordentlich. In vivo sind sie noch unordentlicher, weil Verteilung, Metabolismus, Gewebeanreicherung und Speziesunterschiede verändern, welche Ziele relevant sind. Ein Rezeptoreffekt, der in transfizierten Zellen bei 10 Mikromolar gesehen wird, kann nach normaler oraler Gabe irrelevant sein, während ein schwächer wirkender In-vitro-Effekt wichtig werden kann, wenn die Verbindung sich in lipidreichem Hirngewebe anreichert oder aktive Metaboliten beitragen. Das ist einer der Gründe, warum Schlagzeilen über „den Serotoninrezeptor, den CBD trifft“, den Daten oft vorauslaufen.
Ein weiterer Grund ist Schaltkreisbiologie. Angst entsteht nicht aus einem Rezeptor. Sie ergibt sich aus Interaktionen zwischen Amygdala, bed nucleus of the stria terminalis, medialem präfrontalem Kortex, Hippocampus, Hypothalamus und Hirnstammkernen einschließlich des dorsalen Raphe. CBD scheint die Aktivität in diesem Netzwerk zu verschieben. Ein Teil davon beruht wahrscheinlich auf 5-HT1A. Ein Teil kann TRPV1 betreffen, das bei höheren Dosen anxiolytische Effekte eher entgegenwirken kann. Ein Teil kann FAAH-bezogene Veränderungen des Anandamid-Tonus betreffen, wobei die Frage einer humanen FAAH-Hemmung durch CBD bei therapeutischer Exposition umstritten ist. Ein Teil kann entzündliche oder autonome Effekte widerspiegeln, die wiederum das Erleben von Angst beeinflussen. Sobald diese Netzwerkperspektive übernommen wird, sieht das Scheitern einer Ein-Label-Erklärung nicht mehr wie eine Schwäche aus, sondern wie eine realistische Beschreibung der Pharmakologie.
Auch hierhin entwickelt sich die Arzneimittelforschung. Die Medizinalchemie-Ära interessiert sich weniger dafür, ob eine Verbindung „wie THC“ ist, sondern dafür, Zielkombinationen zu definieren und gewünschte Effekte von Intoxikation zu trennen. Diese Logik zeigt sich in Arbeiten weit jenseits des Serotonins, von der struktur-basierten CB2-Suche im 2016er Journal of Medicinal Chemistry-Paper „Library Docking for Cannabinoid-2 Receptor Ligands“ bis zu neueren Bemühungen, Analgesie von zentraler Beeinträchtigung zu trennen. Sie zeigt sich auch in unternehmensnahen Anxiolyse-Programmen. 2025 sagte MIRA Pharmaceuticals in einer Nasdaq-Pressemitteilung, sein Kandidat MIRA-55 zeige einen „differentiated mechanism of action“ und „anxiolytic activity relative to THC“ in präklinischen Daten. Die Evidenzstufe muss hier klar bleiben: präklinisch, vom Unternehmen berichtet, kein klinischer Beweis. Dennoch ist das Signal als Markt- und Forschungsindikator bedeutsam. Firmen suchen aktiv nach cannabinoid-inspirierten Wirkstoffen, die beruhigen können, ohne wie THC zu wirken, und serotonerge Mechanismen gehören zu dieser Suche.
Der öffentliche Gesundheitskontext macht dies zu mehr als einem akademischen Streit. 2025 erklärte HHS, dass 7-hydroxymitragynine „poses an imminent hazard to public safety“ sei, als es DEA-Maßnahmen gegen gefährliche verstärkte 7-OH-Produkte unterstützte. Unterschiedliche chemische Modifikationen erzeugen unterschiedliche Zielprofile und unterschiedliche Risiken. Dieselbe Lehre gilt für den Cannabinoid-Bereich. Wird ein Produkt als austauschbar mit vertrauten Pflanzen-Cannabinoiden behandelt, nur weil es sich in der Nähe von THC oder CBD anhört, wird die Pharmakologie eingeebnet und die Sicherheitsbewertung leidet.
Wo landet die Evidenz also? 5-HT1A ist der am besten unterstützte serotonerge Mechanismus für die beruhigenden Effekte von CBD, aber die stärkste Aussage, die die Daten derzeit tragen, lautet nicht „CBD ist ein Serotonin-Agonist“. Sie ist enger und besser begründbar: CBD erzeugt häufig anxiolytikähnliche und stressdämpfende Effekte, die teilweise von 5-HT1A-Signalgebung abhängen, während die genaue Art der Beteiligung je nach Assay, Dosis, Gewebe und Schaltkreis-Kontext variiert. Das mag weniger ordentlich sein als ein Ein-Rezeptor-Slogan. Es kommt der Wahrheit jedoch viel näher.
Jenseits der angeforderten Liste: Natriumkanäle und andere nichtkanonische Zielstrukturen, die die Schmerzdebatte bereits verändern
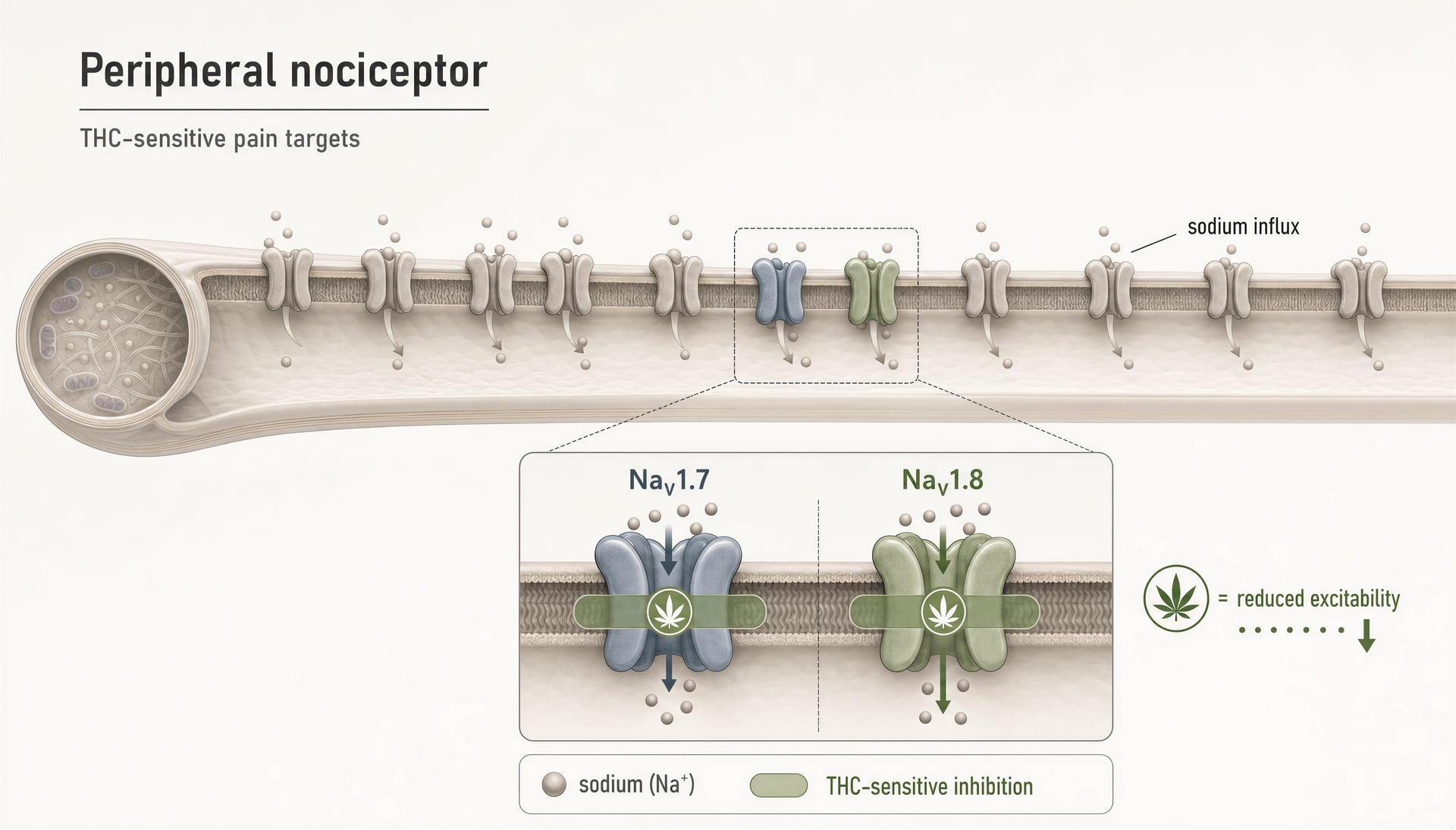
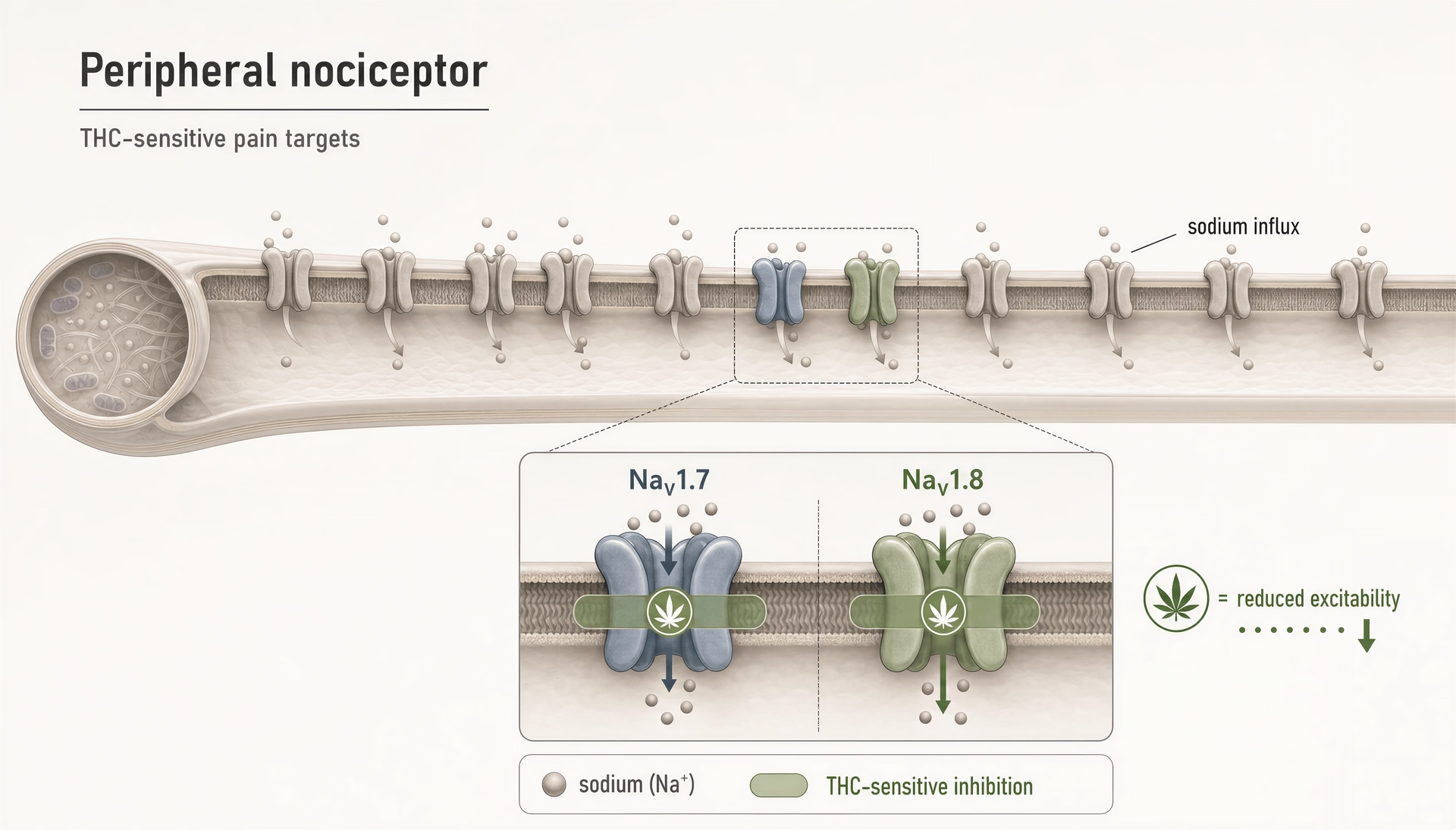
Jahrelang blieb die öffentliche Diskussion über die schmerzbezogene Cannabinoid-Pharmakologie in einer Zwei-Rezeptor-Geschichte stecken: CB1 erklärt psychoaktive Effekte, CB2 erklärt immunologische Effekte, und alles andere gilt als sekundär. Diese Darstellung ist inzwischen zu klein. Selbst im engeren Schmerzfeld berühren Cannabinoide nicht nur TRP-Kanäle, PPARs, Orphan-GPCRs oder serotonerge Signalwege. Sie interagieren auch mit spannungsabhängigen Natriumkanälen, die im Zentrum der Nozizeptor-Erregbarkeit stehen. Das ist wichtig, weil NaV1.7 und NaV1.8 keine peripheren Randbemerkungen sind; sie gehören zu den am meisten untersuchten molekularen Toren der Schmerzsignalgebung in klein-durchmessrigen sensorischen Neuronen.
Der Wandel ist mehr als akademisch. Arzneimittelentwickler suchen seit Jahren nach Möglichkeiten, die Schmerzleitung auf Ebene peripherer Nerven zu blockieren, ohne die Sedierung, Intoxikation, Gedächtnisbeeinträchtigung und Missbrauchsgefahr zu reproduzieren, die mit starker zentraler CB1-Aktivierung verbunden sind. Wenn ein Cannabinoid oder ein cannabinoid-abgeleitetes Gerüst die Feuerrate von Nozizeptoren durch Wirkung auf NaV-Kanäle außerhalb des Gehirns dämpfen kann, eröffnet das eine ganz andere therapeutische Logik. Die Frage verschiebt sich von „Wie stark trifft es CB1?“ zu „Wo wirkt es, bei welcher Konzentration und in welchem Gewebe?“
Diese breitere Zielkarte passt auch zum regulatorischen Moment. 2025 warnte das U.S. Department of Health and Human Services, dass „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety“ sei, eine Erinnerung daran, dass kleine chemische Veränderungen sehr unterschiedliche Pharmakologie- und Sicherheitsprofile erzeugen können. Die Cannabinoid-Politik ist dieser grundlegenden Tatsache oft hinterhergelaufen. Alle intoxikationsnahen Verbindungen so zu behandeln, als unterschieden sie sich nur in Quelle oder THC-Äquivalenz, verfehlt den Punkt. Zielstruktur-Pharmakologie ist das, was Wirkung, Risiko und Wirkstoffpotenzial vorhersagt.
{kind=link}
{kind=link}
{kind=link}
THC hemmt periphere Nozizeptoren durch die Wirkung auf die Natriumkanäle NaV1.7 und NaV1.8.Preliminary evidence
THC an peripheren nozizeptiven NaV1.7- und NaV1.8-Kanälen
Der direkteste Grund, warum Natriumkanäle jetzt in jede ernsthafte Cannabinoid-Karte gehören, ist der Bericht von 2025 aus der Hebrew University of Jerusalem, wonach THC periphere Nozizeptoren hemmt, indem es auf „NaV1.7 and NaV1.8 nociceptive sodium channels“ abzielt. Das erweitert das Vokabular des Feldes erheblich. NaV1.7 und NaV1.8 sind in peripheren schmerzempfindlichen Neuronen stark exprimiert, und ihre Rolle in der menschlichen Schmerzbiologie ist nicht spekulativ. NaV1.7-Verlust-of-Function-Mutationen können zu angeborener Schmerzunempfindlichkeit führen; Gain-of-Function-Mutationen können schwere Schmerzsyndrome verursachen. NaV1.8 ist ebenfalls mit entzündlichen und neuropathischen Schmerzzuständen verknüpft, weil er das repetitive Feuern von Nozizeptoren unterstützt, insbesondere unter depolarisierten Bedingungen.
Wenn also gezeigt wird, dass THC diese Kanäle hemmt, gehört der Befund nicht in die Schublade „verschiedene Off-Target-Effekte“. Er weist auf einen Mechanismus hin, der die Erregbarkeit von Schmerzfasern direkt reduzieren könnte, noch bevor Signale das Rückenmark oder das Gehirn erreichen.
Das ist eine andere mechanistische Klasse als die bekannteren Cannabinoidgeschichten. TRPV1, das in Arbeiten anerkannt wurde, die zu David Julius’ Anteil am Nobelpreis 2021 beitrugen, kann von mehreren Cannabinoiden, einschließlich CBD und CBG, aktiviert oder desensibilisiert werden, wobei die Wirkung stark von Dosis und Zeitverlauf abhängt. PPAR-gamma-Signalgebung wurde für antiinflammatorische und metabolische Effekte herangezogen, oft mit der Komplikation, dass intrazelluläre Anreicherung und Metaboliten ebenso wichtig sein können wie die Ausgangsverbindungen. GPR55 bleibt so umstritten, dass „CB3“ immer noch eher Slogan als gesicherte Wissenschaft ist. Serotonin-Bezüge, insbesondere 5-HT1A, helfen, Teile des anxiolytischen Profils von CBD zu erklären, aber die Schaltkreise sind kontextabhängig und oft indirekt. Natriumkanalhemmung ist weniger glamourös. Sie ist für Schmerz jedoch potenziell praktischer.
Ein wichtiger Punkt hier ist pharmakologische Promiskuität. Cannabinoide sind technisch oft „dirty“ Liganden: Sie binden mehrere Ziele mit unterschiedlichen Affinitäten und funktionellen Konsequenzen. Das ist kein Mangel der Wissenschaft; das ist die Wissenschaft. THC mag weiterhin vor allem für seine zentrale CB1-Agonistik bekannt sein, doch das hebt seine Fähigkeit nicht auf, unter den richtigen Bedingungen periphere Ionenkanäle zu modulieren. Die eigentliche Frage lautet, ob diese Bedingungen in vivo so erreichbar sind, dass sie Patienten mehr nutzen als schaden. Der Befund der Hebrew University sagt, dass dies zumindest plausibel genug ist, um ernsthafte Arzneimittelentwicklungs-Aufmerksamkeit zu verdienen.
Ein cannabinoides Analgetikum kann Schmerzen lindern, ohne über periphere oder Nicht-CB1-Mechanismen ein High zu erzeugen.Preliminary evidence
Periphere Analgesie ohne zentrale Intoxikation
Hier wird das Schmerzfeld interessant. Ein Cannabinoid-Mechanismus, der die periphere Nozizeptor-Feuerung reduziert, könnte zumindest theoretisch Analgesie von der kognitiven Beeinträchtigung trennen, die gewöhnlich mit zentraler CB1-Aktivierung verbunden ist. Diese Unterscheidung steht im Zentrum der aktuellen Translation, nicht am Rand.
Der 2026-ScienceDaily-Bericht brachte die Idee in einfacher Sprache auf den Punkt: Forschende identifizierten „a cannabis compound that relieves pain without the high“. Diese Formulierung sollte sorgfältig gelesen werden. Es handelt sich um ein Forschungssignal, nicht um eine etablierte Therapie, und populäre Zusammenfassungen komprimieren mechanistische Details oft. Dennoch ist die translatioale Bedeutung offensichtlich. Wenn analgetische Aktivität durch periphere Begrenzung, geringe Gehirngängigkeit, selektive Nicht-CB1-Zielbindung oder eine Kombination daraus erzeugt werden kann, dann ist der alte Kompromiss zwischen Schmerzlinderung und Intoxikation nicht naturgegeben. Es ist ein medizinalchemisches Problem.
Dieser Punkt erklärt auch, warum sich das Feld von groben Rezeptorlabels entfernt hat. Das 2016er ACS-Journal-of-Medicinal-Chemistry-Paper „Library Docking for Cannabinoid-2 Receptor Ligands“ spiegelt einen breiteren Wandel zu struktur-basierter Gestaltung wider, statt Cannabinoide als eine pharmakologische Familie mit nur einer nützlichen Variationsachse zu behandeln. Chemiker fragen nun, wie Gerüstform, Lipophilie, Rezeptor-Bias, Gewebeverteilung und Metabolisierung getunt werden können. Das Ziel ist nicht nur stärkere Aktivität. Das Ziel ist selektive Aktivität am richtigen Ort.
Periphere Analgesie ist genau das Endpunkt-Beispiel, bei dem diese Unterschiede zählen. Eine Verbindung, die die Blut-Hirn-Schranke schlecht überwindet, aber NaV1.7 oder NaV1.8 in Nozizeptoren wirksam hemmt, könnte entzündliche oder neuropathische Schmerzen mit weit weniger Intoxikation lindern als THC selbst. Das bleibt ein Ziel, keine klinische Tatsache. Doch die Arbeit der Hebrew University gibt diesem Ziel einen molekularen Anker.[8]Agriculture Improvement Act of 2018. U.S. Congress. Congress.gov, 2018. https://www.congress.gov/bill/115th-congress/house-bill/2/text
Sie schärft auch, wie wir über bereits zirkulierende cannabis-Produkte denken. Die rechtliche Definition von Hanf im 2018 Farm Bill hängt an einem Delta-9-THC-Gehalt von „not more than 0.3 percent on a dry weight basis“. Diese Zahl ist regulatorisch, nicht pharmakologisch. Sie sagt nichts über Natriumkanäle, TRP-Aktivierung, 5-HT1A-Signalgebung, aktive Metaboliten oder Gewebeexposition aus. Dasselbe gilt für neuere verstärkte oder halbsynthetische Intoxikanzien. Sicherheit lässt sich nicht aus Herkunftsgeschichten ableiten. Sie muss aus Zielen, Konzentrationen und realer Pharmakokinetik abgeleitet werden.
Warum diese Befunde für zukünftige Cannabinoid-Medikamente wichtig sind
Die stärkste Implikation der NaV1.7/NaV1.8-Geschichte ist, dass zukünftige Cannabinoid-Medikamente gerade dadurch erfolgreich sein könnten, dass sie im populären Sinn weniger „cannabinoidartig“ sind. Das heißt, die nützlichen Nachkommen der cannabis-Chemie müssen nicht zwangsläufig Arzneimittel sein, die gerauchtes THC breit nachahmen. Es können Verbindungen sein, die einen Teil des Gerüsts übernehmen, zentrale CB1-Signalgebung vermeiden und stattdessen periphere Ionenkanäle oder gemischte nichtkanonische Zielstrukturen adressieren.
Diese Möglichkeit passt bereits zum breiteren Evidenzbild. CBD ist für Krampferkrankungen zugelassen, nicht für routinemäßige Analgesie, und selbst dort wird seine Pharmakologie nicht sauber durch CB1 oder CB2 erklärt. Die FDA-Kennzeichnung für Cannabidiol-Orallösung gibt an, dass sie für Anfälle im Zusammenhang mit Lennox-Gastaut-Syndrom, Dravet-Syndrom oder tuberösem Sklerose-Komplex bei Patienten ab 1 Jahr indiziert ist. Mit anderen Worten: Das einzige größere derzeit breit genutzte FDA-zugelassene Cannabinoid-Medikament widersetzt sich bereits der simplen Rezeptorerzählung. Das Schmerzfeld holt zu derselben Lehre auf.
Erste Unternehmensprogramme weisen in dieselbe Richtung, auch wenn sie Vorsicht verlangen. 2025 berichtete MIRA Pharmaceuticals präklinische Daten, wonach MIRA-55 einen „differentiated mechanism of action“ und „anxiolytic activity relative to THC“ gezeigt habe. Pressemitteilungen von Unternehmen sind keine neutrale Evidenz, und präklinische Signale scheitern oft. Dennoch zeigen sie, wohin die Medizinalchemie geht: weg von ungerichteter THC-Nachahmung und hin zu mechanismengeformtem Design.
Für Schmerz könnten Natriumkanäle zu einem der folgenreichsten Zweige dieser Designstrategie werden. Nicht der einzige Zweig. TRPV1, TRPA1, PPAR-gamma, GPR55, adenosinbezogene Wege und serotonerge Modulation bleiben im Bild. Aber NaV1.7 und NaV1.8 bieten etwas besonders Attraktives: eine direkte Verbindung zum elektrischen Verhalten peripherer Schmerzfasern. Das macht sie in einem Feld voller indirekter Erklärungen ungewöhnlich konkrete Ziele.
Das Ergebnis ist eine klarere Art, Cannabinoide und Schmerz zu denken. Nicht CB1 gegen CB2. Nicht pflanzlich gegen synthetisch. Und nicht „High“ gegen „medizinisch“, als wären das molekulare Kategorien. Die bessere Unterscheidung ist: zentrale Intoxikationsmechanismen versus peripher nützliche Mechanismen. Der Befund der Hebrew University ordnet THC selbst auf beiden Seiten dieser Linie ein. Genau deshalb ist er wichtig.
| Cannabinoid | Im Artikel hervorgehobene Nicht-CB1/CB2-Ziele | Wichtigster Vorbehalt |
|---|---|---|
| CBD | TRPV1, TRPA1, TRPM8, 5-HT1A, PPAR-gamma, GPR55, adenosinbezogene Signalisierung | Viele Signale stammen aus assay-abhängigen und oft mikromolaren Studien |
| THC | TRPV2, berichtete TRPV1/TRPA1-Interaktionen, NaV1.7, NaV1.8 | Zentrale CB1-Effekte können dominieren und andere Mechanismen verschleiern |
| CBG | TRPA1, TRPV1, TRPM8, alpha-2-adrenerge, 5-HT1A-bezogene Interaktionen | Die Übertragung von in-vitro-Konzentrationen auf die Exposition beim Menschen ist unsicher |
| CBC | TRPA1, TRPV-Familienkanäle | Evidenz beim Menschen ist dünn |
| THCV | Nicht-CB1-Möglichkeiten einschließlich TRP- und metabolischer Signalisierung | Dosis und Kontext können das scheinbare Verhalten verändern |
| CBDA / THCA | 5-HT1A-bezogene, TRP- und enzymbezogene Wirkungen, die in präklinischen Arbeiten diskutiert werden | Stabilität, Decarboxylierung und Exposition erschweren die Interpretation |
Wie sich spezifische Cannabinoide unterscheiden, wenn man nicht mehr nur nach CB1 und CB2 fragt
Sobald man CB1 und CB2 nicht länger als die ganze Geschichte behandelt, wirkt das vertraute Cannabinoid-Repertoire weit weniger ordentlich. Diese Moleküle sind keine sauberen Schlüssel für zwei Schlösser. Sie sind lipophile, konzentrationssensitive Verbindungen, die Ionenkanäle, nukleäre Rezeptoren, Transportprozesse, Orphan-GPCRs und in einigen Fällen spannungsabhängige Natriumkanäle treffen können. Das ist wichtig, weil Schmerz, Entzündung, Krampfkontrolle, Angst und Nebenwirkungen oft schlecht zu einfachen Etiketten wie „CB1-Agonist“ oder „CB2-Agonist“ passen.
CBD hat diesen Wandel stärker erzwungen als jedes andere Cannabinoid. Seine geringe klassische Cannabinoid-Rezeptor-Wirksamkeit machte das alte Rahmenmodell schwer verteidigbar, insbesondere nachdem eine FDA-zugelassene Cannabidiol-Orallösung Indikationen für Anfälle im Zusammenhang mit Lennox-Gastaut-Syndrom, Dravet-Syndrom und tuberösem Sklerose-Komplex bei Patienten ab 1 Jahr erhielt. Ein klinisch nützliches Cannabinoid mit begrenzter CB1-ähnlicher Intoxikation war ein Problem für den Rezeptor-Reduktionismus. Forscher mussten anderswo suchen.
Das breitere Feld zog nach. Arbeiten zu TRP-Kanälen stützten sich auf sensorische Biologie, die auf höchster Ebene anerkannt wurde, als der Nobelpreis für Physiologie oder Medizin 2021 an David Julius und Ardem Patapoutian für Entdeckungen von Rezeptoren für Temperatur und Berührung ging. Cannabinoid-Pharmakologie berührt diese Biologie direkt: TRPV1, TRPA1 und TRPM8 tauchen immer wieder auf, weil viele Phytocannabinoide sie bei experimentell relevanten Konzentrationen aktivieren, hemmen oder desensibilisieren können. Das heißt nicht, dass jedes Assay-Ergebnis einen menschlichen Effekt vorhersagt. Es heißt aber, dass die alte Vorstellung, „echte“ Cannabinoid-Wirkung beginne und ende bei CB1/CB2, falsch ist.
CBD: der Prototyp nicht-CB1/CB2-Komplexität
CBD ist das beste Beispiel dafür, warum Ziel-Promiskuität wichtig ist. Es hat im Vergleich zu THC eine niedrige Affinität und geringe Wirksamkeit an CB1 und CB2, tut biologisch aber eindeutig etwas Wichtiges. Die Lücke zwischen diesen beiden Tatsachen führte zu einer Reihe von Arbeits-Hypothesen, manche stärker als andere.
TRP-Kanäle gehörten zu den ersten ernsthaften Alternativen. CBD aktiviert TRPV1 in heterologen Systemen, und TRPV1 ist keine obskure Nebenroute; er ist ein zentraler Kanal für Nozizeption und entzündlichen Schmerz. Aktivierung kann kontraintuitiv klingen, wenn das therapeutische Ziel Analgesie ist, aber wiederholte oder anhaltende TRPV1-Aktivierung führt oft zu Desensibilisierung und damit zu geringerer späterer Erregbarkeit. Das ist einer der Gründe, warum TRP-Pharmakologie auf dem Papier widersprüchlich wirken kann. Eine Verbindung kann zunächst aktivieren und das System später beruhigen. CBD zeigt auch Wirkungen an TRPA1 und kann TRPM8 in einigen Modellen hemmen, was es pharmakologisch breiter macht als die übliche öffentliche Zusammenfassung als „nicht-intoxikierendes Cannabinoid“.
Die Serotonin-Geschichte ist umstrittener, aber weiterhin wichtig. Eine große präklinische Literatur verbindet CBD mit 5-HT1A-bezogenen Effekten, insbesondere in Angst-, Stress- und Übelkeitsmodellen. Die sauberste Aussage ist nicht, dass CBD einfach ein hochaffiner 5-HT1A-Agonist in dem Sinn ist, wie man buspironartige Pharmakologie diskutiert, sondern dass 5-HT1A-Signalgebung oft zu den Wirkungen von CBD in vivo beiträgt, teils durch partielle Agonistik, teils durch allosterische oder circuit-level Mechanismen, die noch ungeklärt sind. Diese Unterscheidung ist wichtig. Zu viele Zusammenfassungen machen aus „bezieht 5-HT1A ein“ schlicht „wirkt über Serotonin“. Die Daten stützen diese Vereinfachung nicht.
PPAR-gamma ist ein weiterer wiederkehrender Kandidat, und hier ist intrazelluläre Chemie entscheidend. PPARs sind nukleäre Rezeptoren, daher kann ein lipophiles Molekül, das sich in Membranen und Zellen verteilt, sie auf eine Weise beeinflussen, die ein Oberflächen-Rezeptormodell verfehlt. CBD wurde in Zellsystemen als PPAR-gamma-aktivierend beschrieben, und PPAR-gamma-Signalgebung hat plausible Bezüge zu Entzündung, Lipidstoffwechsel, Fibrose und Neuroinflammation. Es gibt jedoch einen Haken: Einige PPAR-bezogene Effekte können Metaboliten, längere Exposition oder indirekte Veränderungen endogener Lipidmediatoren widerspiegeln statt einer direkten Rezeptorbelegung. Die Pharmakologie ist real genug, um ernsthaft untersucht zu werden, aber als Schlagwort schwächer als als Mechanismus.
Adenosin-Signalgebung geriet ins Bild, weil CBD in einigen Systemen die Aktivität von Nukleosidtransportern hemmt, was möglicherweise den extrazellulären Adenosin-Tonus erhöht und indirekt antiinflammatorische A2A-abhängige Wege beeinflusst. Auch das ist keine ordentliche Rezeptorbindungs-Pharmakologie. Es ist Transport-Pharmakologie und Gewebekontext. Wenn das unordentlicher klingt als „CBD trifft CB2“, dann ist es das. Es ist auch plausibler.
Dann ist da GPR55, oft als mutmaßlicher „CB3“ gehandelt. Dieses Etikett ist weiterhin zu selbstsicher. CBD kann die GPR55-vermittelte Signalgebung in mehreren experimentellen Systemen antagonisieren oder anderweitig modulieren, und GPR55 wurde mit Erregbarkeit, Knochenbiologie, Entzündung und krebsbezogenen Wegen in Verbindung gebracht. Doch die endogenen Liganden, die Kopplung der Signalwege und die translationale Relevanz bleiben umstritten. GPR55 ist ein nützlicher Hypothesengenerator, kein gesicherter Ersatz für CB1/CB2.
Das Fazit ist einfach: CBD wurde zum Prototyp der Nicht-CB1/CB2-Cannabinoid-Pharmakologie, weil seine klinisch relevanten Effekte sonst nicht zu erklären waren. Das gilt weiterhin.
CBG, CBC, THCV, saure Cannabinoide und die Pharmakologie der Minor-Cannabinoide
Minor-Cannabinoide werden der Öffentlichkeit oft so verkauft, als habe jedes einen einzelnen Charakterzug. Die Pharmakologie kooperiert nicht.
CBG wird gewöhnlich als schwach an CB1 und CB2 aktiv beschrieben, doch die interessanteren Signale liegen außerhalb dieser Rezeptoren. In einigen Assays interagiert es mit alpha-2-adrenergen und 5-HT1A-Systemen, zeigt Aktivität an TRP-Kanälen und wurde hinsichtlich antiinflammatorischer und analgetischer Effekte untersucht, die sich nicht sauber auf klassische Cannabinoid-Rezeptoraktivierung reduzieren lassen. Es illustriert auch ein wiederkehrendes Problem: mikromolare In-vitro-Aktivität ist leicht zu publizieren und schwer zu translieren. Ein Rezeptortreffer bei 10 Mikromolar kann in einer Schale relevant sein und im menschlichen Plasma nicht, oder er kann nur in Geweben relevant sein, in denen sich die Verbindung anreichert.
CBC ist im Vergleich zum Hype um es lange untercharakterisiert gewesen. Es scheint TRPA1- und TRPV-Familienkanäle überzeugender anzusprechen als CB1, und einige Studien deuten auf antiinflammatorische oder analgetikaähnliche Effekte bei Tieren hin. Es gibt auch Interesse an der Wechselwirkung von CBC mit dem Endocannabinoid-Tonus, einschließlich Effekten, die Anandamid-Signalgebung indirekt verändern könnten. Aber „CBC wirkt über TRP-Kanäle“ ist noch ein Ausgangspunkt, keine fertige Antwort. Die humanen Daten sind sehr dünn.
THCV ist ein komplexerer Fall, weil es sich bereits bei CB1 je nach Dosis und Kontext unterschiedlich verhalten kann, oft als neutraler Antagonist oder Ligand mit geringer Wirksamkeit bei manchen Konzentrationen und als stärker agonistisches Molekül bei anderen. Außerhalb von CB1/CB2 wurde THCV mit TRP-Wirkungen und metabolischen Effekten in Verbindung gebracht, mit wiederholtem Interesse an Appetit, glykämischer Kontrolle und Energiebalance. Ein Teil dieser Begeisterung lief den Daten vor Jahren davon. Die bessere Interpretation ist, dass THCV pharmakologisch interessant ist, gerade weil es nicht in das THC-Schema passt, nicht weil irgendein einzelnes sekundäres Ziel sein Profil bereits vollständig erklärt hätte.
Saure Cannabinoide verdienen mehr Aufmerksamkeit als sie erhalten. THCA und CBDA werden oft einfach als „rohe“ Vorstufen behandelt, doch ihre Pharmakologie ist nicht nur inert, bis sie decarboxyliert werden. CBDA besitzt einige der interessantesten Hinweise auf 5-HT1A-bezogene antiemetische Wirkungen in präklinischen Arbeiten, und beide sauren Cannabinoide zeigten in vitro TRP- und enzymbezogene Wirkungen. Ihre geringere Hirngängigkeit im Vergleich zu neutralen Cannabinoiden kann für manche peripheren oder gastrointestinalen Ziele sogar ein Vorteil sein. Das Problem ist die Datenqualität und die Dosisrealität. Viele Behauptungen beruhen auf spärlichen Tierstudien oder Zellassays mit unklarer menschlicher Relevanz.
Hier kollidieren aktuelle Politik und Produktchemie mit der Pharmakologie. Der 2018 Farm Bill definierte Hanf über einen Delta-9-THC-Grenzwert von nicht mehr als 0,3 Prozent auf Trockengewichtsbasis. Diese rechtliche Grenze sagt nichts über GPR55, TRPV1, NaV1.7, Metaboliten oder halbsynthetische Analoga aus. Regulierer reagieren bereits in angrenzenden Bereichen auf diese Diskrepanz. 2025 erklärte HHS, dass „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety“ sei, als es DEA-Maßnahmen gegen verstärkte 7-OH-Produkte unterstützte. Andere Wirkstoffklasse, gleiche Lehre: Modifizierte oder angereicherte Produkte können Sicherheitsprofile haben, die sich stark vom Ausgangsmaterial unterscheiden. Cannabinoide sind von dieser Logik nicht ausgenommen.
Metaboliten, Entourage-Behauptungen und das Chemieproblem
Ein Grund dafür, dass Cannabinoid-Pharmakologie so glitschig wird, ist, dass das Ausgangsmolekül nicht immer die ganze Exposition darstellt. Metaboliten können eine große Rolle spielen. Die Öffentlichkeit lernte dies vor allem über intoxicating THC-Metaboliten, aber das breitere Prinzip gilt für die gesamte Cannabinoid-Wissenschaft. Applikationsweg, First-Pass-Metabolismus, Gewebeverteilung und Speziesunterschiede können verändern, welche Ziele tatsächlich angesprochen werden.
Sogar THC, das eng mit CB1 verbunden ist, ist nicht auf CB1-Biologie beschränkt. 2025 berichteten Forscher der Hebrew University, dass THC periphere Nozizeptoren hemmt, indem es auf die nozizeptiven Natriumkanäle NaV1.7 und NaV1.8 zielt. Das erinnert stark daran, dass Analgesie direkte Effekte auf die Erregbarkeitsmaschinerie einschließen kann, nicht nur GPCR-Signalgebung. Ein 2026 von ScienceDaily hervorgehobener Forschungsbericht trieb dieselbe translationale Idee weiter und beschrieb ein Cannabis-abgeleitetes Molekül, das Schmerz ohne „high“ linderte. Das ist weiterhin Forschungsebene, keine ausgereifte Medizin, weist aber auf eine ernsthafte Arzneimittelentwicklungsstrategie hin: Analgesie von zentraler Intoxikation trennen, indem Nicht-CB1-Ziele, periphere Begrenzung oder beides genutzt werden.
Die Medizinalchemie bewegt sich in diese Richtung. Struktur-basiertes Screening rund um Cannabinoid-Rezeptoren war bereits durch das 2016er Journal of Medicinal Chemistry-Paper „Library Docking for Cannabinoid-2 Receptor Ligands“ gut etabliert, und neuere Bemühungen zielen zunehmend auf differenzierte Mechanismen statt auf generische „THC-like but weaker“-Verbindungen. Eine 2025er MIRA-Pharmaceuticals-Mitteilung, die als Unternehmensbericht und nicht als unabhängige Bestätigung zu behandeln ist, beschrieb MIRA-55 mit einem „differentiated mechanism“ und anxiolytischer Aktivität im Vergleich zu THC. Der entscheidende Punkt ist nicht, dass die Behauptung bewiesen ist. Es ist, dass Arzneimittelentwickler heute Zieltrennung als wichtig ansehen.
Damit kommen wir zum Entourage-Effekt. Als enges wissenschaftliches Konzept ist es plausibel, dass Kombinationen aus Cannabinoiden, Terpenen und Metaboliten Pharmakokinetik verschieben oder additive, antagonistische oder gelegentlich supra-additive Effekte über mehrere Ziele hinweg erzeugen können. Als pauschale Erklärung dafür, warum ein gemischtes Cannabisprodukt angeblich besser wirkt, ist es meist zu vage, um getestet und zu elastisch, um widerlegt zu werden.
Das Chemieproblem ist grundlegend. Eine Mischung aus THC, CBD, CBG, sauren Cannabinoiden, Oxidationsprodukten, Restterpenen und variablen Metaboliten ist keine einzige Intervention. Sie ist viele bewegliche Teile, deren Effekte von Konzentrationsverhältnissen und Zeitverlauf abhängen. Plausible Multi-Target-Pharmakologie existiert. Unbelegte Vereinfachung existiert auch. Das ist nicht dasselbe.
Der bessere Standard ist zielstrukturspezifische Evidenz, verknüpft mit Exposition. Welche Verbindung, bei welcher Konzentration, in welchem Gewebe, mit welchem messbaren Effekt? Sobald diese Fragen gestellt werden, verblasst die Mythologie und die echte Cannabinoid-Geschichte tritt hervor: nicht zwei Rezeptoren, sondern eine gedrängte pharmakologische Karte.
Methoden sind entscheidend: warum das Assay-Design prägt, was wir über die Wirkung von Cannabinoiden glauben
Aussagen über Cannabinoid-Ziele klingen oft sauberer als die Daten dahinter. Ein Paper sagt, CBD „aktiviert TRPV1“, ein anderes nennt es einen „5-HT1A-Agonisten“, ein drittes bezeichnet GPR55 als Cannabinoid-Rezeptor-Kandidaten, und eine Docking-Studie schlägt eine ordentliche Pose in CB2 oder einer Orphan-GPCR-Tasche vor. Diese Aussagen können alle richtungsweisend sein. Sie sind jedoch nicht dieselbe Art von Evidenz.
Dieser Unterschied ist wichtig, weil Cannabinoide fettliebende Moleküle sind, die gern an mehr Orten aktiv erscheinen, als sie in vivo wirklich relevant sind. Das Feld hat dies auf die harte Tour gelernt. Wenn eine Verbindung in Membranen partitioniert, Bilelligenschaften verändert, intrazellulär akkumuliert, aktive Metaboliten bildet oder nur bei 10 bis 50 Mikromolar einen Effekt zeigt, kann sie Zielgeschichten erzeugen, die bei strengeren Bedingungen zusammenbrechen. Für die Cannabis-Pharmakologie sind Methoden kein technisches Randthema. Sie entscheiden, welche Mechanismen überleben.
Bindungsassays, funktionelle Assays und Docking-Studien
Beginnen wir mit der ältesten und saubersten Frage: bindet die Verbindung? In einem Radioliganden-Bindungsassay werden Membranen oder intakte Zellen, die einen Rezeptor exprimieren, mit einem bekannten radioaktiven Liganden plus ansteigenden Konzentrationen des Testmoleküls inkubiert. Wenn das Testmolekül den Radioliganden verdrängt, schätzen die Forschenden die Affinität, oft als Ki angegeben. Das ist nützlich. Es ist auch begrenzt. Bindung sagt nur, dass ein Molekül unter Assaybedingungen eine Stelle besetzen kann; sie sagt nicht, was danach passiert.
Deshalb sind funktionelle Assays wichtiger als Bindung, wenn es um Signalbehauptungen geht. Für TRP-Kanäle wie TRPV1, TRPA1 oder TRPM8 verwenden Forschende oft Calcium-Flux-Assays mit fluoreszierenden Farbstoffen. Wenn die Kanalöffnung Calcium in die Zelle lässt, steigt die Fluoreszenz. Der Reiz liegt auf der Hand: Diese Assays sind skalierbar und können viele Verbindungen schnell vergleichen. Das Problem ist ebenso offensichtlich, wenn man Cannabinoide kennt. Einige Cannabinoide sind fluoreszierend, einige stören Membranen, einige setzen Calcium indirekt aus intrazellulären Speichern frei, und einige lösen nach anfänglicher Aktivierung eine Kanaldesensibilisierung aus. Ein einzelner Peak in einer Calcium-Kurve kann mehrere Mechanismen verdecken.
Patch-Clamp ist langsamer, aber deutlich informativer. Er misst den Ionenstrom direkt. Für Ionenkanäle, besonders Natriumkanäle wie NaV1.7 und NaV1.8, kann Patch-Clamp zeigen, ob ein Wirkstoff Aktivierung, Inaktivierung, Öffnungswahrscheinlichkeit oder Stromdichte verändert. Deshalb ist der 2025er Bericht der Hebrew University über THC an peripheren Nozizeptoren über NaV1.7 und NaV1.8 methodologisch wichtig: Direkte Elektrophysiologie kann einen echten Kanaleffekt von einem vagen zellbasierten Signal unterscheiden. Wenn ein Cannabinoid den Natriumstrom in Nozizeptoren bei Konzentrationen reduziert, die im peripheren Gewebe erreicht werden, hat das für Analgesie mehr mechanistisches Gewicht als ein weiteres lose formuliertes „Rezeptor-Interaktions“-Etikett.
GPCRs und nukleäre Rezeptoren benötigen unterschiedliche Messgrößen. Für GPCRs messen Forschende möglicherweise cAMP, β-Arrestin-Rekrutierung, GTPγS-Bindung, ERK-Phosphorylierung oder Calcium-Signale in konstruierten Zelllinien. Diese sind nicht austauschbar. Ein Cannabinoid kann in einem Signalweg als Agonist, in einem anderen als partieller Agonist und in einem dritten als Antagonist erscheinen. Das ist kein Schlendrian; das ist Signal-Bias. Für PPAR-gamma ist der übliche Ansatz ein Reporterassay, bei dem ein PPAR-empfindlicher Promotor Luciferase antreibt. Das Licht geht hoch, und die Verbindung wird als PPAR-Aktivator bezeichnet. Doch Reporterassays liegen mehrere Schritte von der direkten Zielbindung entfernt. Ein lipophiles Cannabinoid kann Transkription indirekt über Zellstress, Metabolismus oder Veränderungen endogener Lipidmediatoren beeinflussen.
Genexpressionsmessungen liegen noch weiter downstream. Zeigt ein Paper, dass CBD entzündliche Transkripte verändert und der Effekt durch einen PPAR-Antagonisten reduziert wird, ist das suggestiv, nicht endgültig. Der Antagonist kann unvollkommen sein. Die Zellen können Metaboliten bilden. Das Cannabinoid kann Adenosin-Tonus, Calcium-Haushalt, Redoxzustand oder Membranordnung verschieben. Mechanismus durch Ausschluss ist fragil.
Dann gibt es Docking. Docking fragt, ob ein kleines Molekül in eine modellierte oder experimentell bestimmte Bindungstasche passen und günstig scoren kann. Richtig eingesetzt ist es ein Werkzeug der medizinalchemischen Triage. Falsch eingesetzt wird es dekorative Gewissheit. Das 2016er ACS-Journal of Medicinal Chemistry-Paper „Library Docking for Cannabinoid-2 Receptor Ligands“ ist ein guter Einstieg, weil es die Logik von ihrer besten Seite zeigt: Struktur-basiertes Screening kann helfen, Gerüste zu priorisieren, plausible Rezeptor-Ligand-Kontakte zu identifizieren und die Synthese um CB2-Selektivität herum zu lenken. Dafür ist Docking da. Es generiert Hypothesen. Es beweist nicht, dass ein Phytocannabinoid ein Ziel in lebendem Gewebe adressiert, geschweige denn, dass die Interaktion Analgesie, Angst oder Entzündung erklärt.
Speziesunterschiede, Metaboliten und Membraneffekte
Selbst starke In-vitro-Signale können außerhalb der Assayschale versagen, weil die Cannabinoid-Pharmakologie stark kontextabhängig ist. Humane und murine Rezeptoren sind nicht immer funktional identisch. Eine Verbindung, die an murinem TRPA1 oder an rattenbasierten 5-HT1A-bezogenen Readouts aktiv ist, kann an der humanen Orthologform Potenz oder Wirksamkeit verändern. Dieselbe Warnung gilt für Spleißvarianten, Rezeptorreserve und Zellhintergrund. Eine stark überexprimierte Rezeptorlinie kann einen schwachen Liganden wichtig erscheinen lassen.
Der Metabolismus fügt eine weitere Ebene hinzu. Viele Cannabinoide bleiben nicht lange in ihrer Ausgangsform. THC wird zu 11-Hydroxy-THC; andere Strukturen bilden oxidierte oder konjugierte Metaboliten, die sich im Zielprofil deutlich unterscheiden können. Regulierungsbehörden achten in angrenzenden drogenpolitischen Debatten stärker auf dieses Problem. 2025 erklärte HHS, dass „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety“ sei, was eine breitere Lehre hervorhebt: Verstärkte oder metabolisch begünstigte Intoxikanzien können sich stark vom Ausgangsmolekül unterscheiden. Die cannabis-Wissenschaft hat parallele Probleme mit halbsynthetischen Cannabinoiden, neuen Isomeren und Formulierungen, die die Exposition verschieben. Wer nur das Ausgangsmolekül testet, übersieht möglicherweise die Spezies, die die Wirkung in vivo tatsächlich treibt.
Membranen sind der größte stille Störfaktor. Cannabinoide sind lipophil genug, um sich in Bilayer- und intrazellulären Kompartimenten anzureichern. Das bedeutet, dass die nominelle Bad-Konzentration oft ein schlechter Proxy für die Konzentration am Ziel ist. Eine 10-Mikromolar-Applikation im Zellassay kann eine sehr hohe lokale Membranbelastung erzeugen, die Kanal-Gating oder Rezeptorverhalten unspezifisch verändert. Sie kann auch falsche Negative erzeugen, wenn die freie wässrige Konzentration viel niedriger als erwartet ist, weil das Molekül an Plastik, Serumproteine oder die Membran selbst bindet.
Das ist ein Grund, warum Hochkonzentrationsbehauptungen Skepsis verdienen. Wenn ein Cannabinoid ein Ziel erst oberhalb von 20 oder 30 Mikromolar beeinflusst, sollte die erste Frage lauten, ob das eine physiologisch sinnvolle Interaktion oder ein membranvermitteltes Artefakt ist. TRP-Kanäle sind hier besonders anfällig für Übertreibung. Sie sind in einigen Fällen echte cannabinoid-responsive Ziele, aber die Effekte können biphasisch, rasch desensibilisierend und stark konzentrationsabhängig sein. Ein kurzer Calcium-Burst bei hohen Mikromolarwerten bedeutet nicht automatisch einen therapeutisch relevanten Mechanismus.
Von ACS-Docking-Papieren zur realen Pharmakologie
Medizinalchemie lebt von Vereinfachung, Biologie bestraft Übervereinfachung. Das ACS-CB2-Docking-Paper illustriert die nützliche Form der Vereinfachung: mit einer Rezeptorstruktur beginnen, Bibliotheken screenen, Kandidaten priorisieren, Analoga synthetisieren, in Bindungs- und Funktionsassays testen und aus Struktur-Wirkungs-Beziehungen lernen. Diese Abfolge kann echte Medikamente hervorbringen. Sie kann aber auch zeigen, dass die attraktivste in-silico-Pose zu einer Verbindung gehört, die schlechte Permeabilität, instabilen Metabolismus, Signal-Bias oder keine sinnvolle Aktivität in nativen Zellen besitzt.
Die Distanz von der Docking-Pose zur Pharmakologie ist genau der Ort, an dem viele Cannabinoid-Behauptungen scheitern. Ein Docking-Bild von CBD oder CBG in TRPV1, 5-HT1A, GPR55 oder PPAR-gamma ist kein Beweis dafür, dass die Verbindung die relevante Physiologie in Tieren oder Menschen antreibt. Echte Pharmakologie braucht Konvergenz. Idealerweise bedeutet das direkte Zielstruktur-Interaktion bei plausiblen Konzentrationen, funktionelle Effekte in nativen Systemen, Loss-of-Function-Evidenz mittels Antagonisten oder Knockouts, pharmakokinetische Unterstützung und einen Bezug zu Verhalten oder klinischem Ergebnis.
Wenn diese Ebenen übereinstimmen, wird die Geschichte schnell stärker. Der aktuelle Forschungsimpuls, Analgesie von Intoxikation zu trennen, beruht auf genau dieser Logik. Der 2026 von ScienceDaily beschriebene Bericht über „a cannabis compound that relieves pain without the high“ ist interessant, gerade weil er über grobe THC-Äquivalenz hinaus und auf zielselektive oder peripher begrenzte Mechanismen verweist. Das gilt auch für die Natriumkanal-Arbeit zu THC. Dasselbe gilt für Bemühungen, selektive Liganden durch Docking und strukturgeleitetes Design zu bauen. Das Muster ist konsistent, auch wenn einzelne Behauptungen gekürzt werden müssen.
Leser sollten dieselbe Disziplin von akademischen Cannabinoid-Papieren verlangen. Fragen Sie, welcher Assay verwendet wurde. Fragen Sie, ob die aktive Konzentration realistisch ist. Fragen Sie, ob der Effekt Elektrophysiologie, Antagonisten, Knockouts, Metabolismusstudien und Speziesübertragung übersteht. Fragen Sie vor allem, ob die Zielbehauptung den Organismus erklärt, nicht nur die Platte. So wird Nicht-CB1/CB2-Cannabinoid-Pharmakologie von einer Sammlung interessanter Hinweise zu echtem Mechanismus.
Evidenzstufen: von der Zellschale bis zur Klinik
Die Literatur zu Nicht-CB1/CB2-Zielen ist reich, weil Cannabinoide chemisch promiskuitiv sind. Ein einziges Molekül kann je nach Konzentration, Gewebe und Metabolitenprofil TRPV1, 5-HT1A-gekoppelte Signalgebung, PPAR-gamma, GPR55, Adenosin-Tonus und spannungsabhängige Natriumkanäle berühren. Das ergibt interessante Mechanismus-Paper. Es macht noch keine bewiesene Medizin daraus. Wenn es eine Regel gibt, die dieses Feld ehrlich hält, dann diese: Mit jeder Stufe auf der Evidenzleiter verschwinden viele der Behauptungen, die darunter überzeugend wirkten.
Was präklinische Evidenz beweisen kann und was nicht
Am unteren Ende der Leiter stehen Bindungsassays, Kanalaufzeichnungen, Reporter-Systeme und Zellkulturen. Diese Methoden sind unverzichtbar. Mit ihnen lernten Forschende, dass die Cannabinoid-Pharmakologie weit über die klassischen Rezeptoren hinausgeht, und deshalb wirken rezeptor-reduktionistische cannabis-Erklärungen heute altmodisch. Der Nobelpreis 2021 würdigte David Julius und Ardem Patapoutian „for their discoveries of receptors for temperature and touch“, eine Erinnerung daran, dass TRP-Biologie kein Randwissen ist; sie liegt nahe am Zentrum moderner Schmerzforschung. Wenn Cannabinoide TRPV1, TRPA1 oder verwandte Kanäle in einer Schale aktivieren oder desensibilisieren, ist das bedeutsam.
Eine Schale kann jedoch nicht sagen, ob dieselbe Zielbindung bei tolerierbaren menschlichen Dosen auftritt. Viele In-vitro-Effekte von Cannabinoiden erscheinen nur bei mikromolaren Konzentrationen. Plasmawerte nach realer Dosierung können niedriger, flüchtiger oder in Metaboliten verschoben sein, die eine andere Pharmakologie besitzen. CBD ist das klassische Beispiel. Es zeigt wiederholt Wirkungen, die sich schwer nur über CB1 oder CB2 erklären lassen, weshalb TRPV1-, 5-HT1A-, PPAR-gamma-, GPR55- und Adenosin-Wege in der Literatur immer wieder auftauchen. Doch die Tatsache, dass CBD ein Ziel in kultivierten Zellen modulieren kann, beweist nicht, dass dieser Mechanismus ein klinisches Ergebnis bei Epilepsie, Angst, Schmerz oder Entzündung treibt.
Tiermodelle sind der Medizin einen Schritt näher und dennoch weit davon entfernt. Nagetierstudien können zeigen, dass ein Cannabinoid Allodynie reduziert, Entzündungsmarker unterdrückt oder angstähnliches Verhalten verändert. Sie können sogar zielstrukturspezifische Geschichten mit Antagonisten, Knockouts oder peripherer Begrenzung stützen. Der jüngste Bericht der Hebrew University ist ein gutes Beispiel dafür, warum diese Arbeit spannend ist: Forschende berichteten, dass THC periphere Nozizeptoren durch Targeting der nozizeptiven Natriumkanäle NaV1.7 und NaV1.8 hemmt. Dieser Befund widerspricht der simplen Annahme, THC-Analgesie müsse eine CB1-Geschichte plus Intoxikation sein. Hält sich der Effekt, spricht er für eine schmerzrelevante Wirkung auf der Ebene der peripheren Erregbarkeit selbst.
Selbst starke Tierarbeiten beweisen jedoch nicht, dass eine Blockade von NaV1.7/NaV1.8 mit einem Cannabinoid zu einem sicheren, wirksamen humanen Analgetikum wird. Speziesunterschiede zählen. Dosierung zählt. Applikationsweg zählt. Verhaltensendpunkte bei Mäusen können täuschen. Ein in einem Nervenpräparat oder Formalinassay gedämpftes Schmerzsignal muss nicht in Linderung bei Neuropathie, Arthrose oder postoperativen Schmerzen beim Menschen übersetzt werden. Dieselbe Vorsicht gilt für Unternehmensmeldungen. 2025 sagte MIRA Pharmaceuticals, sein Kandidat MIRA-55 habe in präklinischen Daten einen „differentiated mechanism of action“ und „anxiolytic activity relative to THC“ gezeigt. Das ist als Forschungsevidenz legitim. Es ist keine Behandlungsbehauptung und sollte auch nicht so behandelt werden.
Dieselbe translationale Vorsicht gilt für attraktive Schlagzeilen. ScienceDaily hob 2026 Arbeiten zu „a cannabis compound that relieves pain without the high“ hervor. Vielleicht. Das ist ein nützlicher Weg für das Drug Design, besonders wenn periphere Ziele oder Nicht-CB1-Mechanismen Analgesie von zentraler Intoxikation trennen können. Aber „without the high“ in einem präklinischen Bericht ist eine Hypothese unter Test, keine gesicherte Aussage über menschliche Therapie.
Zugelassene Cannabinoid-Medizin und die Enge aktueller Indikationen
Nun zur Spitze der Leiter: zugelassene Arzneimittel-Evidenz. Hier wird das Feld deutlich enger. Das klarste US-Beispiel ist die Cannabidiol-Orallösung. Die FDA-zugelassene Kennzeichnung gibt an, dass sie „for the treatment of seizures associated with Lennox-Gastaut syndrome, Dravet syndrome, or tuberous sclerosis complex in patients 1 year of age and older“ indiziert ist. Dieser Satz ist informativer als Dutzende vager Wellness-Behauptungen, weil er die Darreichungsform, das Ergebnis, die Krankheiten und das Alter nennt.
Beachten Sie, was das Etikett nicht sagt. Es sagt nicht, dass CBD für Schmerz, generalisierte Angst, Insomnie, entzündliche Darmerkrankungen, Neuroprotektion oder eine breite „Endocannabinoid-Balance“ zugelassen ist. Es bestätigt nicht jeden im präklinischen Schrifttum vorgeschlagenen Mechanismus. Die Zulassung sagt uns, dass ein bestimmtes Cannabidiol-Produkt in kontrollierten Studien Wirksamkeit und akzeptable Sicherheit für bestimmte Krampferkrankungen gezeigt hat. Sie entscheidet nicht darüber, ob TRPV1, GPR55, 5-HT1A, PPAR-gamma, intrazelluläre Calcium-Effekte oder Metabolitenwirkungen der dominierende klinische Mechanismus sind. Mechanismus kann teilweise ungeklärt bleiben, selbst wenn die Wirksamkeit real ist.
Diese Lücke zwischen zugelassener Indikation und Mechanismus-Folklore ist in der Pharmakologie häufig. Viele Medikamente wirkten bei Patienten, bevor ihr vollständiges Zielkartenbild verstanden wurde. Cannabinoide sind darin nicht ungewöhnlich. Ungewöhnlich ist, wie oft breite Behauptungen aus verstreuten Rezeptorfunden rückwärts konstruiert und dann so dargestellt werden, als seien sie bereits klinisch geprüft.
Die Enge aktueller Indikationen ist auch für öffentliche Gesundheitsdiskussionen wichtig. Regulierungsbehörden unterscheiden zunehmend vertraute Pflanzen-Cannabinoide von veränderten, verstärkten oder halbsynthetischen Intoxikanzien mit anderen Risikoprofilen. 2025 erklärte HHS, dass „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety“ sei, als es DEA-Maßnahmen gegen verstärkte 7-OH-Produkte unterstützte. Diese Aussage betraf kratombezogene Produkte, nicht cannabis, aber die politische Lehre überträgt sich sauber: Sobald Hersteller potente psychoaktive Moleküle anreichern, modifizieren oder synthetisieren, wird Kurzform-Denken auf Basis des Pflanzenursprungs unzuverlässig. Zielstruktur-Pharmakologie wird dann sehr wichtig.
Warum Mechanismusgeschichten oft klinische Daten überholen
Sie überholen sie, weil Mechanismusgeschichten schnell, anschaulich und publizierbar sind. Klinische Beweise sind langsam und teuer. Ein Zellpaper kann in einem monatelangen Projekt zeigen, dass ein Cannabinoid TRPA1 aktiviert, GPR55 antagonisiert oder 5-HT1A-Signalgebung verändert. Eine überzeugende randomisierte Studie zu chronischen Schmerzen kann Jahre dauern und dennoch scheitern, weil die Effektgröße klein ist, Nebenwirkungen die Dosierung begrenzen oder das präklinische Ziel in Patienten nie sinnvoll erreicht wurde.
Cannabinoid-Chemie lädt auch zur Überinterpretation ein. Strukturell verwandte Verbindungen können sehr unterschiedliche Zielprofile haben, und der Metabolismus kann das Bild nach der Gabe erneut verändern. Der Applikationsweg verschiebt die Geschichte wieder. Orale Gabe erzeugt First-Pass-Metaboliten; Inhalation verändert die Kinetik; topische oder periphere Formulierungen können lokale Ziele gegenüber zentralen bevorzugen. Selbst die rechtliche Kategorie „Hanf“ sagt pharmakologisch wenig aus. Der 2018 Farm Bill zog die Grenze bei Delta-9-THC „not more than 0.3 percent on a dry weight basis“, einer gesetzlichen Schwelle, nicht einer biologischen.
Die richtige Lesart der Nicht-CB1/CB2-Evidenz ist daher weder Ablehnung noch Hype. Die präklinische Literatur zeigt tatsächlich, dass Cannabinoide an mehr als CB1 und CB2 wirken. Insbesondere für Schmerz verdienen TRP-Kanäle und Natriumkanäle ernsthafte Aufmerksamkeit. Bei CBD sind nichtkanonische Ziele keine optionalen Fußnoten; sie sind wahrscheinlich zentral für die Erklärung seines Profils. Aber Plausibilität ist nicht Wirksamkeit, Zielbindung ist kein Patientennutzen, und die Zulassung eines Cannabinoid-Medikaments für eine kleine Gruppe von Krampferkrankungen bestätigt nicht die viel größere Wolke an Behauptungen, die aus Rezeptordiagrammen, Mausverhalten und Zellschalenresultaten konstruiert wurden.
Sicherheit, Regulierung und warum Off-Target-Pharmakologie für die öffentliche Gesundheit wichtig ist
Probleme der öffentlichen Gesundheit folgen nicht sauber Rezeptordiagrammen. Ein Cannabinoid kann pflanzlich, aus Hanf gewonnen, halbsynthetisch oder vollsynthetisch sein und dennoch Risiken erzeugen, die durch sein Etikett, seine Herkunft oder seine rechtliche Kategorie schlecht vorhergesagt werden. Das ist die praktische Bedeutung von Off-Target-Pharmakologie. Sobald ein Molekül TRP-Kanäle, Serotoninrezeptoren, PPARs, GPR55-ähnliche Signalstrukturen, Natriumkanäle und andere Nicht-CB1/CB2-Ziele berührt, kann sich das Sicherheitsprofil auf eine Weise verschieben, die für Vergiftungsüberwachung, Produktstandards, Regeln zum Fahren unter Einfluss und Abhängigkeitsrisiko relevant ist.
Der Fehler ist nicht nur wissenschaftlich. Er ist regulatorisch. Die Cannabis-Politik hat „Cannabinoid“ oft so behandelt, als sei das bereits eine Erklärung.
Verstärkte Intoxikanzien und die Lehre aus 7-OH
Die klarste aktuelle Warnung kommt nicht einmal von einem klassischen cannabis-Bestandteil. 2025 erklärte das U.S. Department of Health and Human Services, dass „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety“ sei, als es DEA-Maßnahmen gegen verstärkte 7-OH-Produkte unterstützte. Dieser Satz ist wichtig, weil er zeigt, dass Bundesgesundheitsbehörden eine Linie zwischen vertrauter botanischer Exposition und konzentrierten oder chemisch manipulierten Intoxikanzien ziehen, die sich in realen Populationen sehr anders verhalten können.
Die politische Lehre überträgt sich direkt auf Cannabinoide. Eine breite Kategoriebezeichnung wie „hemp-derived“, „plant-based“ oder sogar „cannabinoid-like“ sagt Regulierern praktisch nichts darüber, welche Ziele eine Verbindung bei nutzungsrelevanten Konzentrationen anspricht. Sie sagt Verbrauchern auch fast nichts über Potenz, Wirkungseintritt, Dauer, Interaktionsrisiko oder Missbrauchsgefahr.
Verstärkte 7-OH-Produkte wurden zu einem öffentlichen Gesundheitsproblem, weil sich die Chemie änderte und damit die Exposition. Wenn ein Markt von einer natürlichen Spurensubstanz zu einem konzentrierten Wirkstoff übergeht, hört Pharmakologie auf, eine Triviafrage zu sein, und wird zur zentralen Sicherheitsfrage. Dasselbe Muster tauchte bereits bei halbsynthetischen und strukturell veränderten Cannabinoiden auf, die im Fahrwasser der Hanflegalität nach dem 2018 Farm Bill verkauft werden, der Hanf über einen Delta-9-THC-Gehalt von „not more than 0.3 percent on a dry weight basis“ definierte. Diese Trockengewichtsschwelle ist eine Pflanzen-Definition, kein Pharmakologie-Standard. Sie filtert weder TRP-Aktivität, Natriumkanalblockade, 5-HT1A-Signalgebung, GPR55-Effekte noch Metaboliten mit längerer Persistenz und anderer zentralnervöser Penetration heraus.
Deshalb sind Off-Target-Effekte keine obskure Fußnote. Sie sind ein Weg, auf dem Produkte, die öffentlich als cannabisnah vermarktet werden, unerwartete Schäden erzeugen können. Eine Substanz, die öffentlich als „THC-like but legal“ eingeordnet wird, mag sich in der intrinsischen Wirksamkeit an CB1 unterscheiden, aber sie kann sich auch auf viel weniger sichtbaren Ebenen unterscheiden: stärkere kardiovaskuläre Stimulation, größeres prokonvulsives oder anxiogenes Potenzial, schwerere Dysphorie, ungewöhnliche Sedierung oder ein Toxizitätsprofil, das stärker vom Metabolismus als vom Ausgangswirkstoff allein geprägt ist. Diese Möglichkeiten sind nicht abstrakt hypothetisch; genau deshalb reagieren Gesundheitsbehörden auf verstärkte Intoxikanzien anders als auf breite Pflanzenkategorien.
Eine vernünftige Regulierung beginnt mit Fragen auf Zielstrukturebene. Welche Rezeptoren und Kanäle werden angesprochen? Bei welchen Konzentrationen? In welchen Geweben? Was sind die Hauptmetaboliten? Gibt es Hinweise auf Natriumkanalhemmung, die Nozizeption oder kardiale Leitung verändern könnte? Ist TRPV1-Aktivierung wahrscheinlich, Schmerzsignalgebung zu desensibilisieren, oder eher Reizung und Symptomverschlechterung bei niedriger oder kurzer Exposition? Das sind schwierigere Fragen als „ist es ein Cannabinoid“, aber die richtigen.
Die Grenzen des THC-Äquivalenz-Denkens
THC-Äquivalenz ist verführerisch, weil sie Gesetz, Besteuerung, Kennzeichnung und Beeinträchtigungsdebatten vereinfacht. Sie ist jedoch oft falsch. Zwei Verbindungen können eine gewisse Intoxikation erzeugen und sich dennoch stark in Angstneigung, psychotomimetischem Potenzial, analgetischer Wirkung, Herzfrequenzantwort, Emesis-Kontrolle, Toleranzentwicklung und Entzugslast unterscheiden, weil sie nicht dieselbe breitere Zielkarte teilen.
Selbst THC selbst ist pharmakologisch nicht mit CB1 und CB2 erschöpft. 2025 berichteten Forscher der Hebrew University, dass THC periphere Nozizeptoren hemmt, indem es auf die nozizeptiven Natriumkanäle NaV1.7 und NaV1.8 zielt. Dieser Befund widerspricht dem groben Storytelling „THC wirkt hier, CBD dort“. Wenn ein kanonisches psychoaktives Cannabinoid direkt spannungsabhängige Natriumkanäle beeinflussen kann, die für Schmerz relevant sind, dann lassen sich Sicherheit und Wirksamkeit nicht allein aus dem Cannabinoid-Branding ableiten. Dosis, Weg, Verteilung und Gewebeexposition werden entscheidend.
Dasselbe zeigt sich aus der entgegengesetzten Richtung bei CBD. Seine zugelassene orale Lösung ist von der FDA für Anfälle im Zusammenhang mit Lennox-Gastaut-Syndrom, Dravet-Syndrom oder tuberösem Sklerose-Komplex bei Patienten ab 1 Jahr indiziert – eine klinische Tatsache, die sich nie gut durch CB1-Agonismus erklären ließ, weil CBD kein klassisch intoxizierendes CB1-Molekül ist. Sein Profil hat Forscher seit langem zu anderen Mechanismen gedrängt, einschließlich TRPV1, 5-HT1A, adenosinbezogener Signalgebung und PPAR-gamma. Nicht alle diese Mechanismen sind beim Menschen gleich gut etabliert, aber zusammen machen sie einen Punkt unvermeidlich: Cannabinoid-Effekte sitzen oft in einem Netzwerk, nicht an einem einzelnen Rezeptorschalter.
Diese Netzwerkperspektive erklärt auch, warum Intoxikation ein schlechtes Hauptmaß für die öffentliche Sicherheit ist. Ein Produkt kann weniger intoxizierend als THC sein und dennoch bedenkliche Arzneimittelinteraktionen, Lebeenzym-Effekte, kardiovaskuläre Belastung, Panikreaktionen oder Sedierung verursachen. Es kann auch stärker intoxizieren, ohne berechenbarer zu sein. Die Öffentlichkeit hört oft „schwächer als THC“ oder „stärker als THC“, als sei damit alles geklärt. Ist es nicht. Meist handelt es sich um Aussagen über einen auffälligen Phänotyp, nicht über ein vollständiges toxikologisches Profil.
Die Forschungspipeline bewegt sich bereits über THC-Äquivalenz hinaus. Eine 2025 von MIRA Pharmaceuticals veröffentlichte Nasdaq-Mitteilung beschrieb präklinische Daten für MIRA-55 mit einem „differentiated mechanism of action“ und anxiolytischer Aktivität im Vergleich zu THC. Unternehmensmitteilungen sind schwache Evidenz gegenüber peer-reviewter klinischer Datenlage, daher sollte die Behauptung vorsichtig behandelt werden. Dennoch ist die Richtung real: Medizinalchemie versucht, gewünschte Effekte von zentraler Intoxikation zu trennen, indem die Zielbindung verändert wird. Dieselbe translatorische Logik zeigt sich in einem 2026 von ScienceDaily berichteten Arbeitsstand über „a cannabis compound that relieves pain without the high“. Forschungsergebnisse sind kein klinischer Beweis, doch sie unterstreichen einen zentralen regulatorischen Punkt. Wenn sich nützliche Effekte von der Intoxikation trennen lassen, dann lassen sich auch Schäden von der Intoxikation trennen. Ein Produkt mit wenig High ist nicht automatisch ein Produkt mit geringem Risiko.
Warum neue Cannabinoide zielstrukturelle Prüfung verlangen
Neue Cannabinoide verdienen mehr, nicht weniger, Prüfung, gerade weil sie oft in den Handel gelangen, bevor ihre Pharmakologie beim Menschen kartiert ist. Die alte Abkürzung war zu fragen, ob ein Molekül CB1 oder CB2 bindet. Die bessere Frage ist, was es sonst noch tut und ob diese Wirkungen bei realen Dosen nach Inhalation, oraler Aufnahme oder Metabolisierung relevant werden.
Für Schmerz sind TRP-Kanäle und Natriumkanäle offensichtliche Beispiele. David Julius und Ardem Patapoutian erhielten 2021 den Nobelpreis für Physiologie oder Medizin für Entdeckungen von Rezeptoren für Temperatur und Berührung, eine Erinnerung daran, dass somatosensorische Biologie auf Zielstrukturen beruht, die Cannabinoide außerhalb der klassischen Cannabinoid-Rezeptoren beeinflussen können. TRPV1-Aktivierung kann über Desensibilisierung zur Analgesie beitragen, aber auch Reizung erzeugen und ist stark konzentrationsabhängig. Ein Regulator, der nur nach CB1-Aktivität sucht, würde diese gesamte Risiko-Nutzen-Achse übersehen.
Für die psychiatrische Sicherheit ist Serotonin-Signalgebung relevant. CBD wird wiederholt als 5-HT1A-verknüpfter Anxiolysekandidat diskutiert, aber Serotonin-Beteiligung kann indirekt und kontextabhängig sein. Diese Unsicherheit ist kein Grund, das Ziel zu ignorieren; sie ist ein Grund, es sorgfältig zu untersuchen, bevor Produkte als harmlos normalisiert werden. Das gilt ebenso für GPR55, der trotz anhaltender Debatte gelegentlich als „CB3“-Kandidat bezeichnet wird. Ein umstrittenes Ziel ist dennoch ein Ziel, das Calcium-Signalgebung, Erregbarkeit und Entzündungsreaktionen beeinflussen kann.
Für metabolische und entzündliche Effekte verkomplizieren intrazelluläre Ziele wie PPAR-gamma das Bild weiter. Nukleäre Rezeptorsignalgebung sieht nicht wie ein schnelles Intoxikationsereignis aus, kann aber für chronische Exposition, Fettgewebeverteilung, transkriptionelle Veränderungen und Wechselwirkungen mit anderen Krankheitszuständen wichtig sein. Öffentliche Gesundheitsbotschaften, die sich daran orientieren, ob etwas „high“ macht, übersehen diese langsameren, leiseren Risiken.
Hier brauchen Regulierung, Toxikologie und Verbraucherkommunikation molekulare Zielkompetenz. Nicht jeder In-vitro-Treffer ist klinisch relevant. Speziesunterschiede sind real. Metaboliten können dominieren. Aber Behörden sollten Zielstruktur-Panels, Metabolitencharakterisierung, Konzentrations-Wirkungs-Daten und beim Menschen auf Beeinträchtigung bezogene Studien verlangen, bevor sie annehmen, ein neues Cannabinoid könne wie ein anderes reguliert werden. Das 2016er ACS--Journal of Medicinal Chemistry-Paper „Library Docking for Cannabinoid-2 Receptor Ligands“ spiegelt wider, wie Arzneimittelforschung heute arbeitet: struktur-basiertes Design, Selektivitätsengineering und explizite Aufmerksamkeit für rezeptorbasierte Unterschiede. Die Regulierung sollte aufhören zu tun, als sei der Markt einfacher, als die Medizinalchemie es längst weiß.
Die öffentliche Gesundheit kann sich keinen Rezeptor-Reduktionismus leisten. „Cannabinoid“ ist ein Ausgangsetikett. Es ist kein Sicherheitsurteil.
Arzneimittelforschung: Cannabinoide und cannabinoid-inspirierte Moleküle für Nicht-CB1/CB2-Zielstrukturen designen
Die Arzneimittelforschung rund um Cannabinoide ist weit über die alte Frage hinausgegangen, ob ein Molekül „CB1-aktiv“ oder „CB2-aktiv“ ist. Diese Vereinfachung war immer wackelig, weil viele cannabinoidbezogene Verbindungen pharmakologisch unordentlich sind: Sie treffen Ionenkanäle, GPCRs außerhalb des kanonischen Duos, intrazelluläre nukleäre Rezeptoren, Transportprozesse und Metabolisierungsenzyme, oft mit unterschiedlichen Wirkungen je nach Konzentration, Gewebe und Applikationsweg. Für Medizinalchemiker ist dieses Chaos nicht nur ein Problem. Es ist auch eine Chance.
Das zentrale Designziel ist klar genug: analgetische, antiinflammatorische oder anxiolytische Effekte erhalten und zugleich die Belastungen reduzieren, die mit starker CB1-Aktivierung im Gehirn verbunden sind. Diese Belastungen sind nicht abstrakt. Sedierung, kognitive Beeinträchtigung, Intoxikation, Missbrauchspotenzial und dosislimitierende psychiatrische Effekte sind genau der Grund, warum „THC-like“ in einem Entwicklungsprogramm oft ein schlechtes Profil ist, selbst wenn THC selbst nützliche Pharmakologie zeigt. Das aktuelle regulatorische Klima verstärkt diesen Punkt. 2025 unterstützte HHS eine Einstufungsmaßnahme, indem es erklärte, dass „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety“ sei – eine Erinnerung daran, dass modifizierte oder verstärkte Intoxikanzien nicht einfach wie vertraute pflanzenabgeleitete Verbindungen funktionieren dürfen, nur weil sie marketingtechnisch in der Nähe liegen. Zielstruktur-Pharmakologie zählt.
Periphere Begrenzung, funktionelle Selektivität und Signal-Bias
Ein Weg aus zentralen Nebenwirkungen ist grob, aber effektiv: Das Medikament soll nicht ins Gehirn gelangen. Periphere Begrenzung kann durch Erhöhung der polaren Oberfläche, Steigerung der Wasserstoffbrücken-Fähigkeit, Anpassung des pKa oder durch ein Substrat für Efflux-Transporter an der Blut-Hirn-Schranke erreicht werden. Die Idee ist in der Cannabinoid-Wissenschaft nicht einzigartig, passt aber ungewöhnlich gut, weil Schmerzsignalgebung oft in peripheren Nozizeptoren, entzündetem Gewebe und dorsalen Wurzelganglien beginnt.
Dort werden Nicht-CB1/CB2-Ziele besonders attraktiv. Ein Bericht der Hebrew University von 2025 argumentierte, dass THC periphere Nozizeptoren durch Targeting der Natriumkanäle NaV1.7 und NaV1.8 hemmt, zweier Haupttreiber nozizeptiver Erregbarkeit. Wenn dieser Mechanismus sich über Systeme hinweg bestätigt, ist das bedeutsam. NaV1.7 gilt seit Langem als Premium-Schmerz-Ziel, weil Verlust-of-Function-Mutationen in SCN9A eine tiefe angeborene Schmerzunempfindlichkeit auslösen können. Ein Cannabinoid-Gerüst, das die Natriumkanalmodulation in der Peripherie erhält und zugleich zentrale CB1-Signalgebung minimiert, wäre nicht einfach nur „weniger intoxizierendes THC“. Es wäre eine andere Art von Analgetikum.
TRP-Kanäle öffnen einen ähnlichen Raum. Die breitere sensorische Biologie wurde auf höchster Ebene anerkannt, als der Nobelpreis 2021 für Physiologie oder Medizin an David Julius und Ardem Patapoutian für Entdeckungen von Rezeptoren für Temperatur und Berührung ging. TRPV1, TRPA1 und verwandte Kanäle stehen eng mit Nozizeption und entzündlicher Signalgebung in Verbindung, und mehrere Phytocannabinoide interagieren mit ihnen konzentrationsabhängig und manchmal paradox: anfängliche Aktivierung kann von Desensibilisierung gefolgt werden, was zur Analgesie beitragen kann. Das macht Medizinalchemie nicht einfacher. Aber es bedeutet auch, dass eine Verbindung nicht ein sauberer orthosterischer On-Off-Ligand des CB-Rezeptors sein muss, um therapeutisch wertvoll zu sein.
Funktionelle Selektivität fügt eine weitere Ebene hinzu. Selbst an CB1 oder CB2 selbst können Liganden eine Signalantwort gegenüber einer anderen bevorzugen und so das Gleichgewicht zwischen G-Protein-Signalwegen, beta-Arrestin-Rekrutierung, Rezeptorinternalisierung und nachgeschalteten Transkriptionsprogrammen verschieben. Vereinfacht gesagt: Zwei Moleküle können beide „CB1 binden“ und sich in lebendem Gewebe dennoch sehr unterschiedlich verhalten. Das 2016er Journal-of-Medicinal-Chemistry-Paper „Library Docking for Cannabinoid-2 Receptor Ligands“ spiegelt wider, wie stark sich die Designstrategie hin zu struktur-basiertem Tuning statt zu groben Rezeptorbezeichnungen verschoben hat. Dieselbe Logik reicht nun über die CB-Rezeptoren hinaus: Chemiker wollen Gerüste, deren Form, Lipophilie und Konformationszwänge sie in Richtung eines schmerzrelevanten oder anxiolytischen Mechanismus lenken, ohne schweres zentrales Signal-Baggage zu erzeugen.
CBD ist der bleibende Beweis dafür, dass ein cannabinoidbezogenes Medikament klinisch relevant sein kann, ohne durch CB1/CB2-Agonismus erklärt zu werden. Die 2024 aktualisierte FDA-Kennzeichnung für Cannabidiol-Orallösung umfasst Anfälle im Zusammenhang mit Lennox-Gastaut-Syndrom, Dravet-Syndrom und tuberösem Sklerose-Komplex bei Patienten ab 1 Jahr. Welche Kombination aus TRPV1-, 5-HT1A-, GPR55-bezogenen, adenosinergen, intrazellulären und Netzwerk-Effekten diese Wirksamkeit letztlich erklärt, ist keine einfache CB1-Geschichte. Arzneimittelforscher haben das erkannt.
MIRA-55 und der Vorstoß zu differenzierten Mechanismen
MIRA-55 ist eine nützliche Fallstudie, nicht weil sie etwas abschließend klärt, sondern weil sie zeigt, wie Unternehmen Cannabinoid-Programme heute rahmen. In einer 2025 über Nasdaq verbreiteten Pressemitteilung sagte MIRA Pharmaceuticals, sein Kandidat habe einen „differentiated mechanism of action“ und „anxiolytic activity relative to THC“ in präklinischer Arbeit gezeigt. Diese Formulierung verrät fast alles über die aktuelle Investoren- und Regulierungssprache. Cannabinoid-inspiriert zu sein reicht nicht mehr. Ein Unternehmen will Distanz zu bloßer THC-Nachahmung, besonders bei Indikationen wie Angst, wo zentrale Nebenwirkungen den Nutzen zunichtemachen können.
Skepsis ist dennoch zwingend. „Differentiated mechanism“ in einer Unternehmensmitteilung ist eine Behauptung, kein Schluss. Es kann veränderte Rezeptorbindung, andere Gewebeverteilung, unterschiedliche Metaboliten, partielle Agonistik, funktionellen Bias, Off-Target-Ionenkanäle oder schlicht ein anderes Verhaltensprofil in einem Tierassay bedeuten. Ohne vollständige Pharmakologie-Panels, Konzentrations-Wirkungs-Kurven, Metabolitenidentifikation, Rezeptorbelegungsdaten und verblindete Replikation ist die Formulierung eher Hypothese als Ergebnis.
Die dahinterstehende Strategie ist dennoch plausibel. Wenn eine Verbindung angstähnliches Verhalten reduzieren und dabei Intoxikation, Gedächtnisstörung oder Lokomotionshemmung im Vergleich zu THC vermindern kann, hat sich die Medizinalchemie wahrscheinlich in einem von vier Punkten verändert: Hirngängigkeit, intrinsische Wirksamkeit an CB1, Ansprache von Nicht-CB-Zielen wie 5-HT1A oder TRP-Kanälen oder Metabolisierung in aktive Spezies mit anderem Zielprofil. Genau auf diesen Achsen konkurrieren moderne Cannabinoid-Programme.
MIRA-55 zeigt auch das Problem der Ziel-Dekonvolution. Cannabinoid-ähnliche Moleküle sind pharmakologisch oft „dirty“. Das ist kein moralisches Urteil, sondern eine mechanistische Warnung. Tritt ein präklinisches anxiolytisches Signal auf, darf man nicht annehmen, dass ein einzelner Rezeptor alles erklärt. Serotonin-Signalgebung kann direkt oder indirekt sein. PPAR-gamma-Effekte können intrazelluläre Akkumulation oder Metaboliten erfordern. GPR55 kann in einem Assay wichtig und in einem anderen marginal wirken. Ein In-vitro-Treffer bei 10 Mikromolar kann irrelevant sein, wenn freie Gehirnkonzentrationen in vivo nie an diese Größenordnung herankommen.
Wie eine realistische Cannabinoid-Pipeline aussieht
Eine realistische Pipeline ist enger und disziplinierter, als die öffentliche Rhetorik suggeriert. Es ist kein Festzug „nicht-psychoaktiver Cannabisverbindungen“ auf dem Weg zur Zulassung. Es ist ein Filter.
Am Anfang stehen Gerüstmodifikation und Triage: klassische Cannabinoide, nichtklassische Cannabinoide, endocannabinoid-inspirierte Lipide und völlig andere Chemotypen, die eine nützliche Eigenschaft der Cannabinoid-Pharmakologie nachahmen, ohne das ganze Paket zu erben. Chemiker verändern Seitenkettenlänge, Ringzwänge, Stereochemie, Heteroatompositionen und metabolische Schwachstellen und testen dann nicht nur CB1 und CB2, sondern TRPV1, TRPA1, NaV-Kanäle, ausgewählte Orphan-GPCRs und serotonergebundene Assays. Treffer durchlaufen dann ADME-Arbeiten, ungebundene Konzentrationsanalysen und Messungen von Gehirn-zu-Plasma-Verhältnissen. Viele sterben dort.
Die Programme, die überleben, teilen sich gewöhnlich in einige glaubwürdige Kategorien. Eine sind peripher begrenzte Analgetika, bei denen der Traum Schmerzlinderung durch Natriumkanäle, TRP-Desensibilisierung, entzündliche Modulation oder periphere Cannabinoid-Signalgebung ohne wesentliche ZNS-Exposition ist. Eine andere sind biased oder niedrigwirksame CB1-Liganden, die therapeutische Signalgebung erhalten und Intoxikation verringern sollen. Eine dritte sind Mischmechanismus-Verbindungen, oft realistischer als target-puristischer Ansatz, bei denen moderate Wirkungen an mehreren Schmerz- oder Angst-Knoten einen „sauberen“, aber klinisch schwachen Liganden schlagen.
Der translationale Haken, der dieses Feld lebendig hält, ist einfach: Die Trennung von Analgesie und High scheint zumindest in Forschungssystemen möglich. Die 2026 von ScienceDaily zusammengefasste Arbeit über „a cannabis compound that relieves pain without the high“ sollte vorsichtig gelesen werden, weil Schlagzeilen die Daten überlaufen, aber das Konzept passt zur breiteren Richtung der Medizinalchemie. Dasselbe gilt für die Natriumkanal-Arbeit zu THC. Dasselbe gilt für Bemühungen, selektive Liganden durch Docking und strukturgeleitetes Design zu entwickeln. Das Muster ist konsistent, auch wenn einzelne Behauptungen gekürzt werden müssen.
Wahrscheinlich sieht die Pipeline kurzfristig nicht nach massenhaftem klinischem Erfolg aus. Ziel-Promiskuität, Metabolitenkomplexität, Speziesunterschiede und Formulierungseffekte zerstören einfache Geschichten immer wieder. Aber das Feld fragt nicht länger, ob Cannabinoide „wirklich“ von CB1 und CB2 handeln. Arzneimittelentwickler haben diese Frage mit ihren Chemiebudgets bereits beantwortet. Sie designen für den Raum jenseits dieser Rezeptoren, weil dort jetzt die beste Chance liegt, Nutzen von Nebenwirkungen zu trennen.
Häufige Missverständnisse und ungelöste Kontroversen
Der größte Fehler in der öffentlichen Cannabinoid-Diskussion ist der Rezeptor-Reduktionismus: der Drang, eine unordentliche Pharmakologie auf ein Schlagzeilen-Ziel zu pressen. Diese Gewohnheit war immer wackelig, und sie wird riskanter, während Regulierer und Arzneimittelentwickler mit Verbindungen konfrontiert sind, die nicht mehr einfach „THC“ oder „CBD“ im einfachen, pflanzenabgeleiteten Sinn sind. 2025 erklärte HHS, dass „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety“ sei, als es DEA-Maßnahmen gegen verstärkte 7-OH-Produkte unterstützte. Andere Wirkstoffklasse, gleiche Lehre: Sobald Chemiker einen intoxikierenden Scaffold verändern, anreichern oder halbsynthetisch herstellen, können alte Annahmen über Rezeptorwirkung und Sicherheit schnell scheitern. Die cannabis-Wissenschaft hat dasselbe Problem. Ein Cannabinoid ist kein magischer Schlüssel für ein Schloss. Es ist gewöhnlich ein promiskuitiver Ligand, dessen reale Effekte von Konzentration, Gewebeexposition, Metabolismus und den Off-Targets abhängen, die auf diesen Ebenen relevant werden.
Gibt es wirklich einen CB3-Rezeptor
Nein, es gibt keinen Konsens über einen CB3-Rezeptor.
Diese Antwort klingt hart, weil die Evidenz sie verlangt. Über die Jahre wurden mehrere Rezeptoren als „CB3“-Kandidaten vorgeschlagen, insbesondere GPR55, manchmal GPR18 und gelegentlich andere Orphan-GPCRs, die in einigen Assays auf Cannabinoid-Liganden oder endocannabinoidbezogene Lipide reagieren. Aber eine vorgeschlagene Zielstruktur ist keine anerkannte Rezeptorklasse. CB1 und CB2 erhielten ihre Namen durch konvergierende Evidenz: Klonierung, reproduzierbare Ligandenpharmakologie, Gewebeverteilung, Signalgebung und breite Replikation. Die angeblichen CB3-Kandidaten haben diese Hürde nie genommen.
GPR55 ist der übliche Verdächtige. Er wird im Gehirn, in Immunzellen, im Darm und im Knochen exprimiert, und einige Cannabinoide interagieren tatsächlich mit ihm. CBD wurde in Zellassays oft als GPR55-Antagonist beschrieben; bestimmte synthetische Cannabinoide zeigen ebenfalls Aktivität. Doch die Pharmakologie ist zwischen Laboren, Liganden und Readouts inkonsistent. Einige Verbindungen wirken in einem Signalassay aktiv und in einem anderen still. Speziesunterschiede verkomplizieren das Bild. Endogene Liganden sind umstritten. Vor allem suggeriert das Etikett „CB3“ einen festen Platz innerhalb der kanonischen Cannabinoid-Rezeptorfamilie, den das Feld ihm nicht gegeben hat.
Das ist wichtig, weil Labels die Evidenz überholen können. Sobald ein Rezeptor einen eingängigen Spitznamen hat, beginnt der Spitzname, Erklärungsarbeit zu leisten, die er nicht verdient hat. Schmerz? CB3. Angst? CB3. Knocheneffekte? CB3. Das ist keine Pharmakologie; das ist Branding. Die vorsichtigere Position lautet, dass GPR55, GPR18, GPR119 und verwandte Rezeptoren Nicht-CB1/CB2-Zielstrukturen mit unterschiedlichem Grad an Evidenz für Modulation durch Cannabinoide oder cannabinoidähnliche Lipide sind. Einige könnten in spezifischen Geweben sehr wichtig sein. Keiner hat den Konsensstatus als „dritter Cannabinoid-Rezeptor“ erreicht.
Die Arzneimittelforschung hat sich entsprechend bewegt. Medizinalchemie-Paper tun nicht so, als würde ein neuer Rezeptorname das System lösen. Das 2016er Journal of Medicinal Chemistry-Paper „Library Docking for Cannabinoid-2 Receptor Ligands“ ist ein guter Marker dafür, wo das Feld tatsächlich steht: struktur-basiertes Design, Rezeptorselektivität, Gerüstengineering und zielgerichtete Optimierung, nicht Mythologie über einen geheimnisvollen CB3, der alles erklären soll. Ebenso betonen Aussagen zu nächsten Generationen cannabinoid-inspirierter Verbindungen heute oft differenzierte Mechanismen statt einfach stärkerer Rezeptoragonistik. MIRA Pharmaceuticals’ präklinische Pressemitteilung von 2025 zu MIRA-55 behauptete explizit einen „differentiated mechanism of action“ und anxiolytische Aktivität im Vergleich zu THC. Das ist Werbematerial, keine gesicherte Wissenschaft, spiegelt aber einen echten Strategiewechsel wider: Nützliche Cannabinoid-Therapeutika könnten daraus entstehen, dass man von grober CB1-Intoxikation weggeht, nicht daraus, dass man einen Alles-erklärenden Rezeptor entdeckt.
Wirkt CBD hauptsächlich über Serotonin
Ebenfalls nein, obwohl Serotonin-Signalgebung Teil der Geschichte ist.
CBD wird in der Popkultur häufig so vermarktet, als sei es im Grunde ein natürliches 5-HT1A-Medikament. Diese Vereinfachung hält sich, weil reale Evidenz dahintersteht. In präklinischen Studien zeigte CBD anxiolytische und stressdämpfende Wirkungen, die in einigen Paradigmen durch 5-HT1A-Antagonisten abgeschwächt werden. Auch humane experimentelle Studien ließen vermuten, dass serotonerge Mechanismen unter bestimmten Bedingungen zu akuten anxiolytischen Effekten beitragen. Aber „trägt bei“ ist nicht „wirkt hauptsächlich über“.
CBD ist pharmakologisch breit. Es hat im Vergleich zu THC eine geringe Affinität zu CB1 und CB2, aber das macht es nicht automatisch zu einem Einziel-Serotonin-Medikament. Über die Literatur hinweg gehören zu den plausiblen Mitverursachern TRPV1, 5-HT1A, GPR55, Adenosin-Signalgebung, PPAR-gamma, intrazelluläres Calcium-Handling, FAAH-bezogener Endocannabinoid-Tonus in manchen Kontexten und Wirkungen von Metaboliten, die nicht zum Ausgangsmolekül passen. Die Konzentration ist hier wichtiger, als viele Erklärtexte zugeben. Ein Rezeptoreffekt, der in vitro bei mikromolaren Werten gesehen wird, muss bei einer Standard- oralen Dosis beim Menschen nicht dominieren, wenn die Absorption variabel und der First-Pass-Metabolismus erheblich ist.
Der klinische Befund spricht gegen Ein-Mechanismus-Behauptungen. Die stärkste FDA-anerkannte Anwendung von gereinigtem CBD ist nicht Angst, sondern Krampferkrankungen: Die Orallösung ist indiziert für Lennox-Gastaut-Syndrom, Dravet-Syndrom und tuberösen Sklerose-Komplex bei Patienten ab 1 Jahr. Diese zugelassene Anwendung sagt bereits etwas aus. Wenn CBD „hauptsächlich Serotonin“ wäre, ließe sich sein reproduzierbarstes therapeutisches Profil schwer mit dem in Einklang bringen, was Kliniker tatsächlich nutzen. Serotonin mag bei angstbezogenen Situationen wichtig sein, insbesondere bei 5-HT1A-verknüpften Antworten, aber die menschliche Pharmakologie von CBD lässt sich nicht auf ein Serotonin-Label reduzieren.
Selbst innerhalb von Angst verschiebt sich der Mechanismus wahrscheinlich mit Dosis und Kontext. TRPV1 ist ein gutes Beispiel. CBD kann TRPV1 aktivieren, und TRP-Signalgebung ist für die sensorische Biologie so zentral, dass David Julius und Ardem Patapoutian 2021 den Nobelpreis für Entdeckungen von Rezeptoren für Temperatur und Berührung erhielten. TRPV1-Effekte sind jedoch nicht linear. Aktivierung kann von Desensibilisierung gefolgt werden; niedrige und hohe Dosen können unterschiedliche Verhaltensausgänge erzeugen. Wenn also jemand sagt, „CBD wirkt über Serotonin“, ist die richtige Antwort: teilweise, manchmal und wahrscheinlich nicht allein.
Kann ein Ziel einen ganzen Pflanzen-Effekt erklären
Nein, kein einzelnes Ziel kann einen Ganzpflanzen-Cannabis-Effekt erklären, und wer es erzwingen will, verdeckt meist mehr, als er klärt.
Ganzpflanzen-Effekte entstehen aus gestapelten Variablen. Beginnen wir mit der Zusammensetzung: THC, CBD, Minor-Cannabinoide wie CBG, CBC, THCV, saure Vorstufen, Oxidationsprodukte und Metaboliten bringen jeweils unterschiedliche Zielprofile mit. Fügt man den Applikationsweg hinzu, verändert sich das Bild erneut. Inhalierte Cannabinoide erreichen das Gehirn schnell; orale Produkte unterliegen dem First-Pass-Metabolismus und erzeugen andere aktive Spezies. Das US-Recht definiert Hanf immer noch über einen Delta-9-THC-Grenzwert von „not more than 0.3 percent on a dry weight basis“, aber diese rechtliche Linie sagt wenig über die Pharmakologie aller anderen Bestandteile der Probe oder über das, was nach dem Metabolismus entsteht.
Dann kommt die Gewebespezifität hinzu. Ein Cannabinoid kann parallel zentrale CB1, periphere TRP-Kanäle, immunologische Signalwege und nukleäre Rezeptoren beeinflussen. Der 2025er Bericht der Hebrew University, wonach THC periphere Nozizeptoren durch Targeting von NaV1.7 und NaV1.8 hemmt, ist ein scharfes Beispiel, weil er die bequeme Gleichung „THC-Effekt=CB1-Effekt“ durchbricht. Wenn sogar THC relevante Natriumkanalwirkungen in Schmerzbahnen hat, wird die Idee schwer verteidigbar, eine Blüte, ein Extrakt oder ein Edible habe einen einzigen Meisterrezeptor. Die 2026 von ScienceDaily hervorgehobene Forschung – ein Cannabis-Molekül, das Schmerz ohne High lindert – weist in dieselbe Richtung, auch wenn sie Forschungsebene bleibt. Analgesie könnte sich durch periphere Begrenzung oder Nicht-CB1-Ziele von zentraler Intoxikation trennen lassen. Dorthin bewegt sich das Feld.
Auch Erwartung und individuelle Biologie spielen eine Rolle. Vorwissen, Angstniveau, Genetik, Geschlecht, Leberenzyme, Schlaf, Entzündung und Begleitmedikationen verändern, wie sich ein Produkt anfühlt und was es physiologisch tut. Terpene können in manchen Fällen beitragen, sollten aber nicht als magische Regisseure des Gesamteffekts behandelt werden. Ihre Konzentrationen sind oft niedrig, und die menschliche Evidenz ist dünner als die Marketingsprache suggeriert.
Die härtere, aber genauere Sicht lautet: Cannabis-Effekte entstehen emergent. Sie ergeben sich aus vielen kleinen Interaktionen, nicht aus einem Schlagwort-Rezeptor. Das macht die Wissenschaft weniger sauber. Es macht sie auch ehrlicher.
Praktische Einordnung für Leser, Kliniker und Forscher
Die praktische Lehre aus der Nicht-CB1/CB2-Cannabinoid-Pharmakologie ist einfach, aber anspruchsvoll: Mechanismusbehauptungen sollten mehr Skepsis, nicht mehr Zustimmung bekommen, sobald ein Paper TRP-Kanäle, PPARs, GPR55, 5-HT1A, adenosinerge Signalgebung oder NaV-Kanäle erwähnt. Cannabinoide sind häufig pharmakologisch promiskuitive Moleküle. Das kann in der Arzneimittelforschung nützlich sein. Es kann Leser jedoch auch dazu verleiten, jeden Rezeptortreffer in einer Schale als Erklärung für einen klinischen Effekt zu behandeln.
Eine gute Regel ist, die Evidenz nach Distanz zum Patienten zu ordnen. Ein Bindungsassay ist der Anfang, nicht das Ende. Zell-Signalstudien kommen danach. Tierarbeit kann die Plausibilität schärfen. Humane experimentelle Pharmakologie zählt mehr. Zugelassene Arzneimittel-Evidenz zählt am meisten, und selbst dann muss das Etikett den Mechanismus nicht vollständig festlegen. Die Cannabidiol-Orallösung ist beispielsweise von der FDA für Anfälle im Zusammenhang mit Lennox-Gastaut-Syndrom, Dravet-Syndrom und tuberösem Sklerose-Komplex bei Patienten ab 1 Jahr zugelassen, doch ihr therapeutisches Profil wird nicht sauber durch CB1 oder CB2 erklärt. Genau diese Lücke ist der Grund, warum Behauptungen über TRPV1, GPR55, 5-HT1A und intrazelluläre Ziele immer wieder auftauchen.
Wie Cannabinoid-Mechanismus-Behauptungen kritisch zu lesen sind
Beginnen Sie mit Spezies und System. Wurde der Effekt in humanem Gewebe, einer Nagetierzelllinie, Xenopus-Oozyten oder überexprimierten Rezeptoren in HEK293-Zellen gezeigt? Das ist nicht austauschbar. GPR55 ist ein Paradebeispiel: Eine Studie kann CBD in einem rekombinanten System als Antagonisten zeigen, während ein anderer Kontext schwache oder variable Signalgebung erzeugt, weil Rezeptorexpression, endogene Lipide und Assay-Design unterschiedlich sind. Das als „den Mechanismus“ zu bezeichnen, ist meist verfrüht.
Dann die Konzentrationsfrage. Hier brechen viele Cannabinoid-Behauptungen zusammen. Ein Paper kann TRPV1-Aktivierung, PPAR-gamma-Transaktivierung oder Natriumkanalhemmung bei mikromolaren Konzentrationen berichten. Gut. Aber erzeugt die diskutierte menschliche Dosis tatsächlich freie Gewebekonzentrationen in dieser Größenordnung? Orale Cannabinoide stehen vor First-Pass-Metabolismus, starker Proteinbindung und ungleichmäßiger Gewebeverteilung. Ein Target-Treffer bei 30 Mikromolar in vitro kann interessante Chemie und irrelevante Bettseiten-Pharmakologie bedeuten. Umgekehrt kann das Gegenteil eintreten: Lokale Gewebeanreicherung, aktive Metaboliten oder Lipidpartitionierung können ein intrazelluläres Ziel plausibler machen, als Plasmawerte allein suggerieren. So oder so: Konzentration ist kein Randthema. Sie ist das Thema.
Direkte Zielmessung ist ebenfalls wichtig. Haben die Forschenden den Effekt tatsächlich mit einem selektiven Antagonisten blockiert, den Rezeptor herunterreguliert oder den Kanalstrom gemessen? Oder wurde der Mechanismus aus Ähnlichkeit mit früherer Literatur abgeleitet? Für TRP-Kanäle, insbesondere TRPV1 und TRPA1, ist das wichtig, weil Aktivierung biphasisch und desensibilisierend sein kann. Eine Verbindung kann zunächst einen Kanal aktivieren und dann die nachgeschaltete Reaktionsfähigkeit senken, was bedeutet, dass „Agonist“ nicht immer sauber mit „mehr Schmerz“ oder „mehr Hitzewahrnehmung“ gleichzusetzen ist. Das ist einer der Gründe, warum der Nobelpreis 2021 an David Julius und Ardem Patapoutian, verliehen „for their discoveries of receptors for temperature and touch“, hier so relevant ist: Somatosensorische Signalgebung ist mechanistisch reich, und einfache Rezeptorlabels versagen oft.
Die NaV-Geschichte zeigt, wie stärkere Evidenz aussieht. Forscher der Hebrew University berichteten 2025, dass THC periphere Nozizeptoren durch Targeting der nozizeptiven Natriumkanäle NaV1.7 und NaV1.8 hemmt. Diese Behauptung ist informativer als vage Aussagen, THC wirke „außerhalb von CB1“, weil sie schmerzrelevante Kanäle benennt, die bereits im Zentrum der Analgesieforschung stehen. Sie rahmt auch eine alte Annahme neu. Selbst ein Molekül, das für zentrale CB1-vermittelte Intoxikation berühmt ist, kann unter den richtigen Bedingungen klinisch relevante Nicht-CB-Wirkungen haben, besonders im peripheren Gewebe.
Leser sollten auch auf konkurrierende Erklärungen achten. Angenommen, eine Studie verbindet CBD über 5-HT1A mit Anxiolyse. Vernünftige Hypothese. Aber wurde Sedierung ausgeschlossen? Wurde CB1-Modulation indirekt bedacht? Trennte das Experiment Rezeptor-Effekte von Veränderungen des Endocannabinoid-Tonus, der Adenosinaufnahme, der Entzündungssignalgebung oder von Erwartung beim Menschen? Multi-Target-Medikamente kündigen in der Regel nicht an, welcher Zielmechanismus in einem bestimmten Modell den größten Anteil hat.
Das aktuelle regulatorische Klima macht diese Skepsis mehr als akademisch. 2025 erklärte HHS, dass „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety“ sei, als es DEA-Maßnahmen gegen verstärkte 7-OH-Produkte unterstützte. Das ist kein cannabis-Fall, aber die Lehre überträgt sich sauber: Regulierer unterscheiden zunehmend vertraute Pflanzenbestandteile von verstärkten, halbsynthetischen oder anderweitig modifizierten Intoxikanzien mit unterschiedlichen Potenz- und Sicherheitsprofilen. Eine Cannabinoid-Politik, die alle Verbindungen wie skaliertem Delta-9-THC behandelt, ist pharmakologisch überholt.
Welche Fragen Kliniker zur Zielrelevanz stellen sollten
Kliniker müssen nicht jeden Orphan-GPCR auswendig kennen, um Cannabinoid-Behauptungen sinnvoll zu bewerten. Sie brauchen eine disziplinierte Checkliste.
Erstens: Wurde der Effekt beim Menschen gezeigt oder nur in Zellen und Tieren? Ein Mausmodell für Entzündung kann die Plausibilität von PPAR-gamma- oder TRPA1-Beteiligung stützen, beweist aber keinen Patientennutzen. Zweitens: Bei welchen Konzentrationen oder Dosen? Wenn ein vorgeschlagenes Ziel nur bei Werten aktiviert wird, die Standard-orale Dosierung nicht erreicht, erklärt dieser Mechanismus vielleicht keine routinemäßigen klinischen Ergebnisse. Drittens: Wurde das Ziel direkt gemessen? Rezeptorbelegung, Elektrophysiologie, Antagonisten-Umkehr oder genetische Störung zählen mehr als narrative Ableitung.
Viertens: Macht der Applikationsweg die Behauptung glaubwürdiger oder weniger glaubwürdig? Inhalation, orale Einnahme, transdermale Gabe und topische Anwendung erzeugen sehr unterschiedliche Expositionsprofile. Ein peripherer analgetischer Mechanismus ist für eine topische oder peripher begrenzte Verbindung plausibler als für ein Molekül, das schnell das Gehirn flutet. Das ist wichtig, wenn das Therapieziel Analgesie ohne Intoxikation ist.
Fünftens: Gibt es konkurrierende Erklärungen, die mit bekannten Nebenwirkungen zusammenhängen? Wenn ein Patient nach einer Cannabinoid-Zubereitung weniger Angst berichtet, ist das ein 5-HT1A-vermittelter anxiolytischer Effekt, weniger Schmerz, unspezifische Sedierung oder Erwartung? Wenn Entzündungsmarker sich verändern, ist PPAR-gamma der wahrscheinliche Treiber oder haben sich breitere metabolische oder immunologische Veränderungen vorgelagert? Mechanismus sollte nicht allein aus Symptombesserung abgeleitet werden.
Für Kliniker verdienen Minor-Cannabinoide dieselbe Vorsicht. CBC, CBG, THCV und saure Cannabinoide werden oft so dargestellt, als besäßen sie jeweils ein stabiles, charakteristisches Zielprofil. Die Literatur rechtfertigt diese Sicherheit noch nicht. Einige sind vielversprechend. Keines sollte als pharmakologisch gesichert gelten, nur weil eine Marke oder ein Social-Media-Thread Zielstrukturspezifität behauptet.
Wohin sich das Feld wahrscheinlich als Nächstes entwickelt
Die glaubwürdigste kurzfristige Richtung ist periphere Analgesie. Der translationale Reiz liegt auf der Hand: Schmerzlinderung von zentraler Intoxikation trennen. Der 2026 von ScienceDaily berichtete Arbeitsstand über „a cannabis compound that relieves pain without the high“ sollte als Forschungssignal, nicht als fertige klinische Antwort gelesen werden, aber er zeigt, wohin die Medizinalchemie zielt. Dasselbe gilt für die NaV1.7/NaV1.8-Arbeit der Hebrew University von 2025: Die Schmerzbiologie drängt die Cannabinoid-Wissenschaft zu peripheren Nerven, Ionenkanälen und gewebeselektivem Zugang.
Antiinflammatorische Liganden an nukleären Rezeptoren sind eine weitere starke Spur. PPAR-gamma-Behauptungen brauchen bessere Target-Engagement-Daten beim Menschen, aber das Konzept ist plausibel genug, um eine ernsthafte Entwicklung zu rechtfertigen, besonders dort, wo metabolische und entzündliche Wege zusammentreffen. Multitarget-Anxiolytika werden ebenfalls wahrscheinlich ein aktives Terrain bleiben. MIRA Pharmaceuticals sagte in einer 2025er Nasdaq-Mitteilung, sein Kandidat MIRA-55 habe in präklinischen Daten einen „differentiated mechanism of action“ und anxiolytische Aktivität im Vergleich zu THC gezeigt. Da dies unternehmensberichtet und präklinisch ist, sollte es vorsichtig behandelt werden. Dennoch spiegelt es einen realen Trend wider: Forscher geben sich nicht mehr damit zufrieden zu fragen, ob ein Kandidat „wie THC“ oder „wie CBD“ ist. Sie wollen definierte Zielprofile.
Dieselbe Verschiebung zeigt sich in der Medizinalchemie. Das 2016er Journal of Medicinal Chemistry-Paper „Library Docking for Cannabinoid-2 Receptor Ligands“ signalisierte einen struktur-basierten Ansatz, der seither nur gewachsen ist: Liganden für spezifische Ziele, spezifische Gewebe und spezifische Signal-Bias bauen. Besser definierte Minor-Cannabinoide werden wahrscheinlich aus dieser Denkweise hervorgehen, nicht aus breiten Behauptungen, dass jedes seltene Cannabinoid eine einzigartige Wellness-Nische habe.
Die praktische Schlussfolgerung ist nüchtern. Beim Lesen von Cannabinoid-Pharmakologie jedes Mal fünf Fragen stellen: Wurde der Effekt beim Menschen oder nur in Zellen gezeigt? Bei welchen Konzentrationen? Wurde das Ziel direkt gemessen? Erreicht die Dosis dieses Ziel in lebendem Gewebe? Gibt es konkurrierende Erklärungen? Die Zukunft der Cannabinoid-Wissenschaft ist nicht weniger spezifische Pharmakologie, sondern mehr.
Quellen
- [1]HHS and FDA support DEA action on dangerous 7-OH products. HHS Press Room, 2025. https://www.hhs.gov/press-room/hhs-fda-support-dea-7-oh-scheduling.html
- [2]EPIDIOLEX (cannabidiol) oral solution label. FDA drug label, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/210365s016lbl.pdf
- [3]Library Docking for Cannabinoid-2 Receptor Ligands. Journal of Medicinal Chemistry, 2016. https://pubs.acs.org/doi/10.1021/acs.jmedchem.6c00835
- [4]MIRA Pharmaceuticals Reports New Preclinical Data Demonstrating MIRA-55's Differentiated Mechanism of Action and Anxiolytic Activity Relative to THC. Nasdaq press release, 2025. https://www.nasdaq.com/press-release/mira-pharmaceuticals-reports-new-preclinical-data-demonstrating-mira-55s
- [5]Psychoactive cannabinoid THC inhibits peripheral nociceptors by targeting NaV1.7 and NaV1.8 nociceptive sodium channels. Research portal summary, 2025. https://cannabinoids.huji.ac.il/publications/psychoactive-cannabinoid-thc-inhibits-peripheral-nociceptors-targeting
- [6]A cannabis compound that relieves pain without the high. ScienceDaily, 2026. https://www.sciencedaily.com/releases/2026/06/260619033343.htm
- [7]The Nobel Prize in Physiology or Medicine 2021. Nobel Prize Press Release, 2021. https://www.nobelprize.org/prizes/medicine/2021/press-release/
- [8]Agriculture Improvement Act of 2018. Congress.gov, 2018. https://www.congress.gov/bill/115th-congress/house-bill/2/text