






Pourquoi CB1 et CB2 comptent en science du cannabis
GPCR Récepteur couplé aux protéines G : un récepteur membranaire qui change de conformation après la liaison d’un ligand et signale via des partenaires intracellulaires tels que les protéines G et les bêta-arrestines.
Ce qui détermine la réponse du récepteur
- Localisation Le tissu, le type cellulaire et la position subcellulaire façonnent la réponse.
- Identité du ligand Les endocannabinoïdes, les phytocannabinoïdes et les ligands synthétiques n’induisent pas des états de signalisation identiques.
- Partenaires disponibles Différentes cellules offrent différentes protéines G, kinases et bêta-arrestines.
- Schéma d’exposition La durée du signal et la stimulation répétée influencent la désensibilisation et l’internalisation.
- Biais de voie Un ligand peut favoriser la signalisation par les protéines G, le recrutement des bêta-arrestines ou d’autres réponses.
Le langage courant a longtemps réduit la biologie des cannabinoid à une distinction simple : CB1 expliquerait le « high », CB2 prendrait en charge l’inflammation quelque part hors du cerveau. Ce cadrage est beaucoup trop simpliste pour être utile. CB1 et CB2 sont des récepteurs couplés aux protéines G, ou GPCRs, et, comme les autres GPCRs, ils ne fonctionnent pas comme de simples interrupteurs marche/arrêt. Ils traduisent les signaux des endocannabinoid produits par l’organisme, des phytocannabinoid issus de Cannabis sativa, et des ligands synthétiques conçus en laboratoire en réponses cellulaires variables. La réponse observée dépend de l’emplacement du récepteur, du ligand qui s’y lie, des partenaires de signalisation disponibles, de la durée de stimulation du récepteur, et du fait que celui-ci soit orienté vers la signalisation par protéines G, le recrutement de β-arrestin, la désensibilisation ou l’internalisation.
Cela compte, car la science du cannabis ne se limite pas à l’intoxication. Elle concerne aussi la douleur chronique, l’épilepsie, la signalisation immunitaire, la neurodégénérescence, les risques psychiatriques, et la raison pour laquelle tant de programmes de médicaments cannabinoid ont paru prometteurs en préclinique avant d’échouer chez l’humain. Les enjeux sont considérables. L’Organisation mondiale de la Santé a estimé que 200 millions de personnes ont utilisé du cannabis en 2019, soit environ 4 % de la population mondiale âgée de 15 à 64 ans. L’épilepsie touche environ 50 millions de personnes dans le monde. La schizophrénie concerne environ 24 millions de personnes. Et pourtant, en 2025, la FDA américaine ne note l’approbation que d’un produit médicamenteux dérivé du cannabis et de trois produits médicamenteux liés au cannabis. Cet écart entre une exposition massive et un nombre limité de thérapeutiques approuvées explique en partie pourquoi la biologie des récepteurs est si importante.
| Caractéristique | CB1 | CB2 |
|---|---|---|
| Abréviation typique | « récepteur cérébral » | « récepteur immunitaire » |
| Correction apportée par l’article | Enrichissement central, mais aussi expression périphérique | Enrichi dans le système immunitaire, mais non sans pertinence pour le cerveau |
| Fonctions exemples mentionnées | Perception, mémoire, contrôle moteur, nociception | Signalisation des cytokines, migration cellulaire, rôles neuro-inflammatoires |
| Interprétation | Dépendant du circuit et de l’état | Dépendant du type cellulaire et de l’état pathologique |
Pourquoi la biologie des récepteurs explique davantage que les étiquettes de la plante
Les étiquettes telles que « indica », « sativa », ou même « THC-dominant » et « CBD-dominant » ne disent qu’une partie de l’histoire, car ce sont les récepteurs, et non les catégories marketing de la plante, qui sont les plus proches du mécanisme. Le Δ9-tétrahydrocannabinol (THC) est un agoniste partiel de CB1 et CB2, mais l’effet en aval du THC n’est pas figé. Dans les neurones corticaux riches en CB1, il peut diminuer la libération de neurotransmetteurs et modifier la perception, la mémoire et le contrôle moteur. Dans les voies sensorielles périphériques, la même famille de récepteurs peut moduler la nociception. Dans les cellules immunitaires, l’activation de CB2 peut modifier la signalisation des cytokines ou la migration cellulaire. Même famille. Résultats différents.
La règle simpliste selon laquelle CB1 serait uniquement cérébral et CB2 uniquement immunitaire est trop rudimentaire pour la biologie actuelle des récepteurs.Strong evidence
La vieille règle simplificatrice — CB1 dans le cerveau, CB2 dans les cellules immunitaires — reposait sur un schéma réel, mais elle a mal vieilli. La distribution est fondée sur des gradients et spécifique des types cellulaires, et non binaire. CB1 est fortement exprimé dans de nombreuses régions du système nerveux central, surtout sur les terminaisons présynaptiques, mais il apparaît aussi dans les tissus périphériques. CB2 est fortement associé à la fonction immunitaire, mais l’idée qu’il serait sans pertinence pour le cerveau n’est plus défendable. Une revue de 2026 dans Frontiers in Behavioral Neuroscience a soutenu que la signalisation de CB2 a suscité un intérêt croissant dans les troubles du système nerveux central, notamment via des mécanismes neuroinflammatoires et neurodégénératifs, et l’a décrite comme « une mise à jour des 3 dernières années ». Cette mise à jour est importante. Si CB2 contribue, dans certaines conditions, à une pathologie centrale, alors les médicaments visant CB2 ne peuvent pas être compris comme de simples outils périphériques.
La structure approfondit encore le tableau. Une revue de 2026 dans Frontiers in Chemical Biology a expliqué que la sélectivité des ligands entre « CB1 et CB2 » découle de différences structurelles au niveau des récepteurs, qui influencent la conformation de liaison, l’efficacité et la régulation du récepteur. En termes simples, de petits changements chimiques peuvent orienter un ligand vers un sous-type de récepteur ou vers une voie de signalisation particulière, ce qui aide à expliquer pourquoi deux cannabinoid qui se ressemblent sur le papier peuvent produire des effets très différents in vivo. Une étude indexée dans PubMed de 2025/2026 sur la sélectivité des sous-types a poussé l’analyse plus loin en montrant que la sélectivité des endocannabinoid est liée à la dynamique conformationnelle des récepteurs plutôt qu’à un modèle rigide de type serrure-clé. Le récepteur bouge. Le ligand stabilise certains états plus que d’autres. La biologie suit ces états.
Des phytocannabinoid à la signalisation des endocannabinoid
{kind=link}
Signalisation rétrograde Un schéma de signalisation synaptique dans lequel une cellule postsynaptique libère un messager qui agit en sens inverse sur des récepteurs présynaptiques.
Séquence de découverte en bref
- 1 Le THC a été isolé et sa structure définie.
- 2 Des sites spécifiques de liaison aux cannabinoïdes ont été démontrés dans le tissu cérébral.
- 3 CB1 a été cloné comme GPCR.
- 4 CB2 a été identifié dans des tissus liés au système immunitaire.
- 5 L’anandamide puis le 2-AG ont établi un système de signalisation endogène.
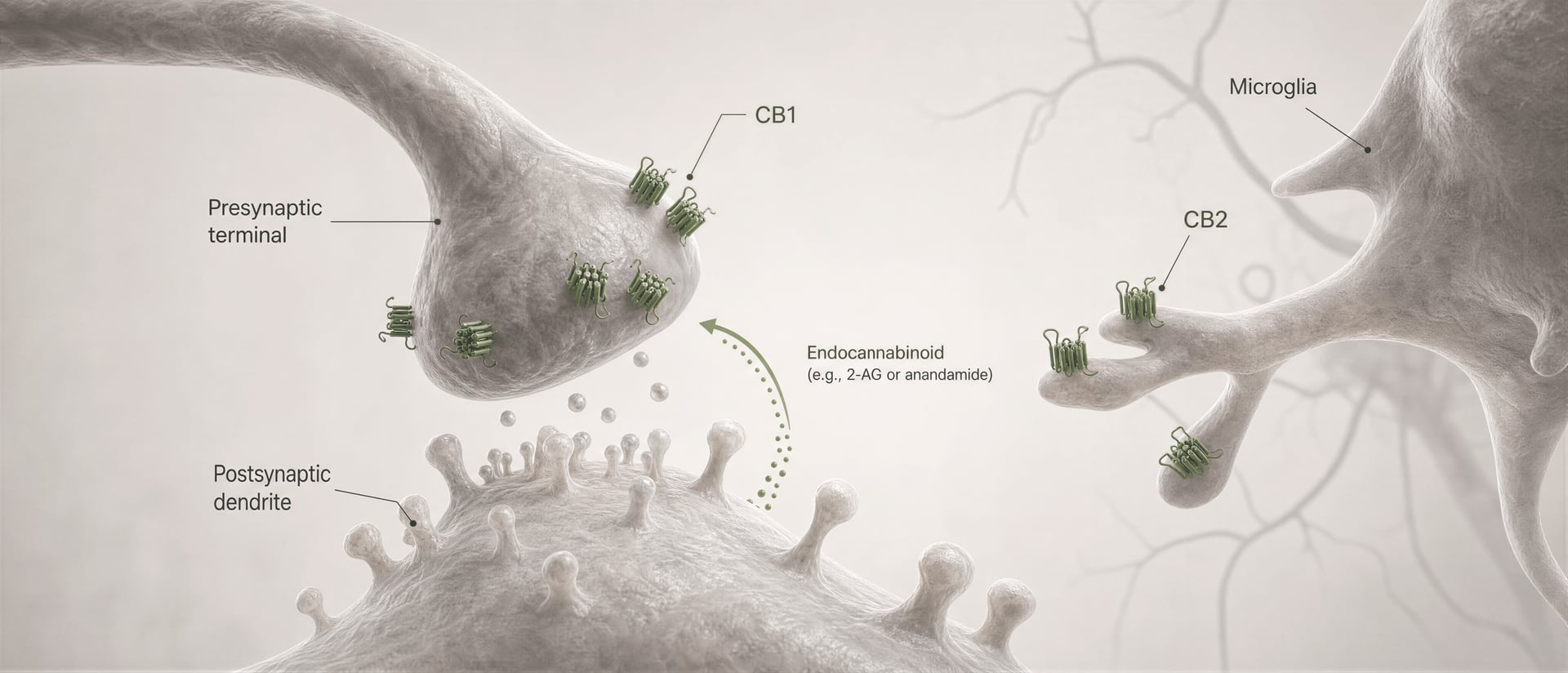
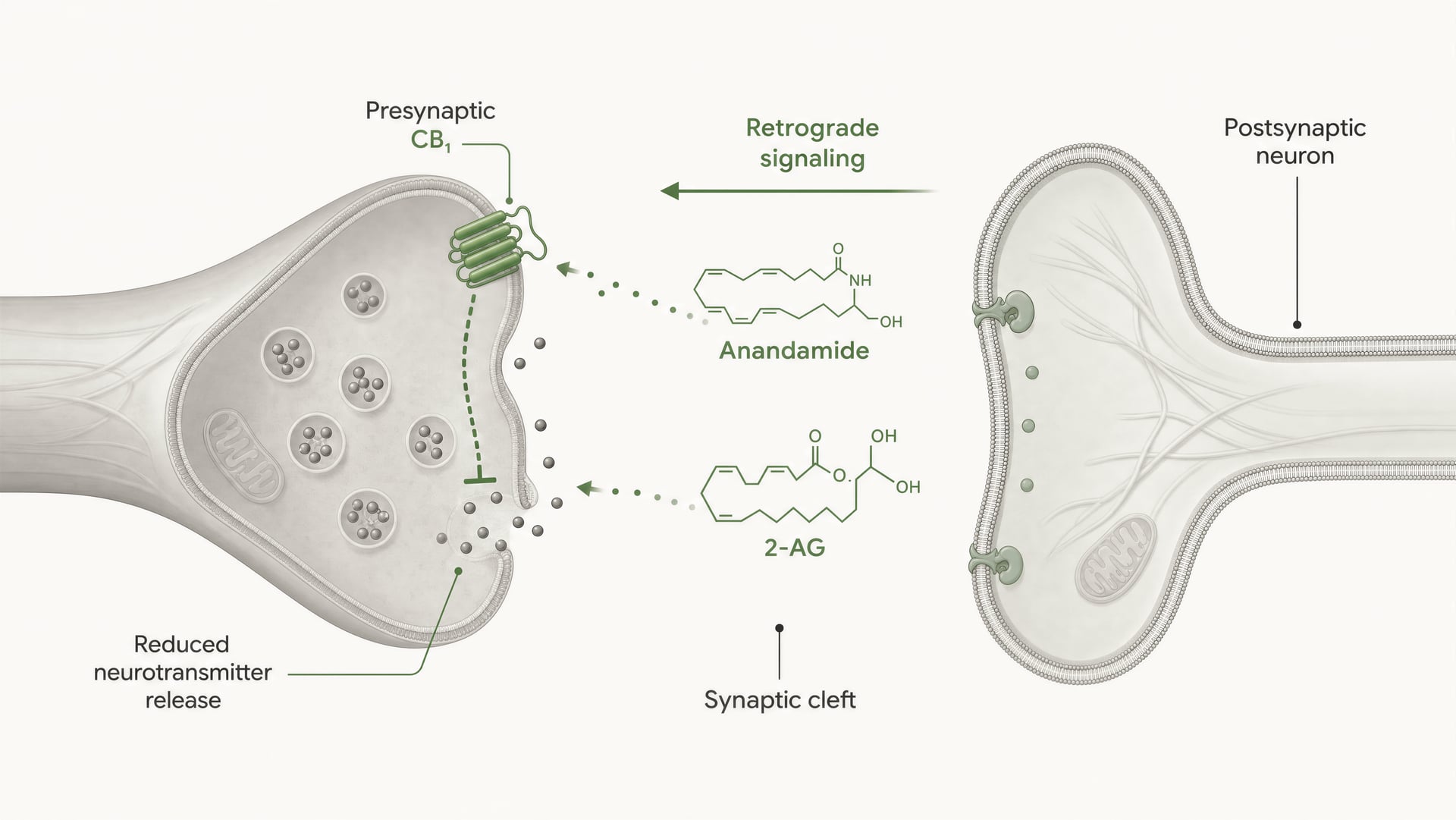
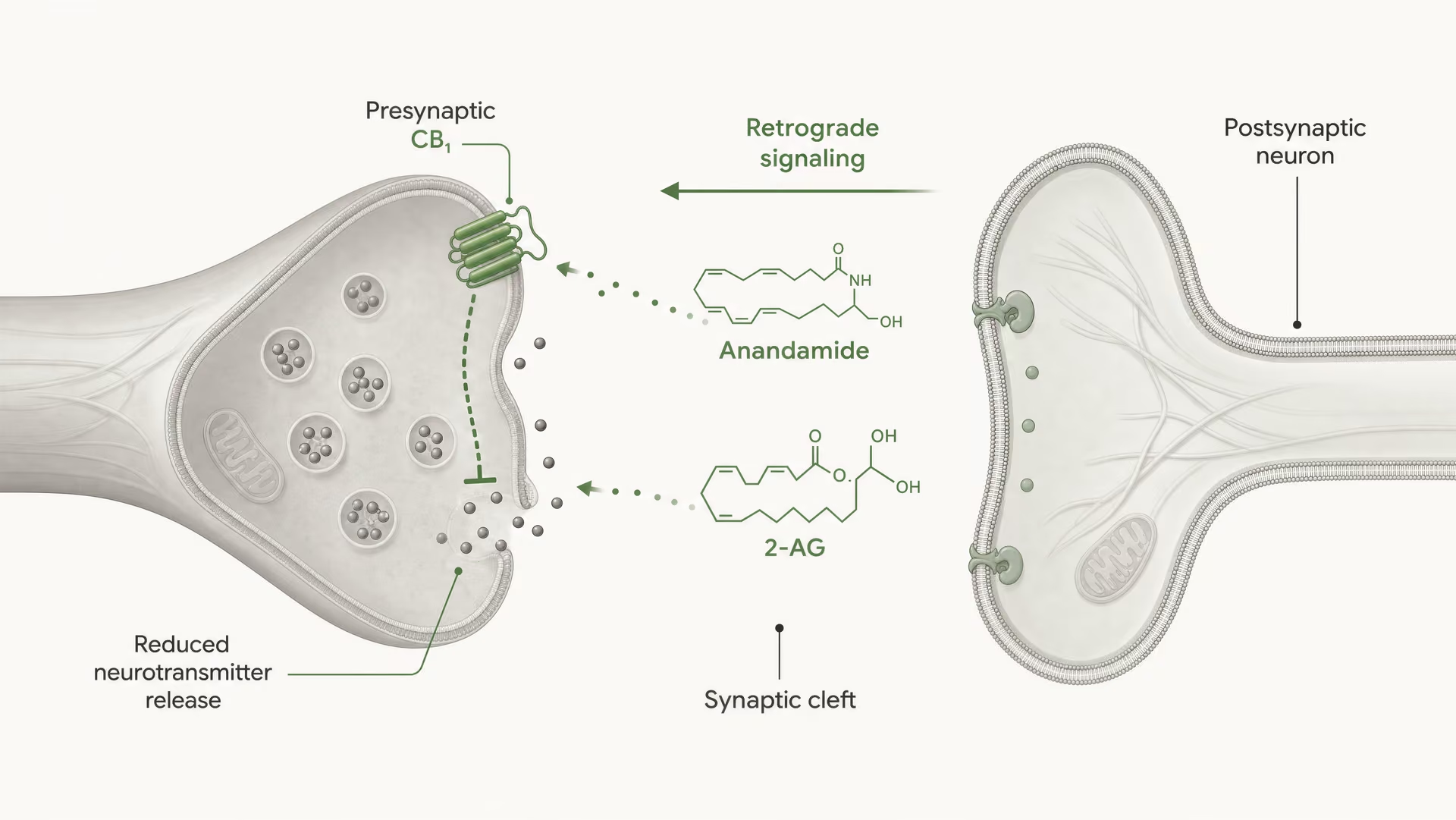
Les récepteurs cannabinoid n’ont pas été découverts parce que l’organisme aurait évolué pour le cannabis. La chronologie a suivi le chemin inverse. Les travaux d’Allyn Howlett et de ses collègues ont joué un rôle central dans la définition de la pharmacologie des récepteurs cannabinoid, et la découverte ultérieure de l’anandamide par Raphael Mechoulam et Lumír Hanuš a contribué à établir que l’être humain produit ses propres molécules de signalisation de type cannabinoid. L’anandamide et le 2-arachidonoylglycérol, généralement abrégé en 2-AG, sont les principaux endocannabinoid. Ils sont produits à la demande, et non stockés dans des vésicules comme les neurotransmetteurs classiques, et ils signalent souvent de manière rétrograde : une cellule postsynaptique génère un endocannabinoid qui traverse la synapse en sens inverse pour activer CB1 présynaptique et réduire la libération ultérieure de neurotransmetteurs.
Cela offre une image fondamentalement différente de « la molécule de weed qui frappe un récepteur ». La signalisation des endocannabinoid est locale, transitoire et étroitement régulée par des enzymes de synthèse et de dégradation. Les phytocannabinoid entrent dans ce système depuis l’extérieur. Les ligands synthétiques peuvent l’activer de façon plus forte ou plus sélective encore. Il en résulte qu’un même récepteur peut être sollicité par une brève impulsion endogène, par un phytocannabinoid oral absorbé lentement, ou par un agoniste synthétique à forte efficacité présentant des risques de sécurité très différents.
Cette différence explique en partie pourquoi l’intoxication ne peut pas être déduite du seul nom du récepteur. Elle dépend de l’efficacité du ligand, de la dose, de la voie d’administration, du moment et du contexte tissulaire. Le THC au niveau de CB1 est bien central dans les effets psychoactifs, mais ce fait ne réduit pas CB1 à un simple « récepteur de la psychoactivité ». Il ne fait pas non plus de CB2 une simple commande anti-inflammatoire. L’article de 2025 dans American Journal of Psychiatry sur la signalisation biaisée de CB1 a précisément formulé ce point plus large en soutenant que des ligands biaisés de CB1 pourraient offrir une stratégie thérapeutique pour la schizophrénie. Cette proposition relie la science du cannabinoid à une idée plus générale des GPCRs : si un ligand favorise des branches de signalisation bénéfiques tout en évitant celles associées à des effets indésirables, l’action du médicament peut être dissociée d’une activation brute du récepteur. Reste à savoir si cette promesse se confirmera en clinique, mais l’argument mécanistique est solide.
Ce que cet article entend par distribution, signalisation et cibles médicamenteuses
Dans cet article, distribution signifie davantage qu’une carte des organes. Cela inclut la densité du récepteur, le type cellulaire, la localisation subcellulaire, l’état pathologique et l’évolution temporelle. Un récepteur exprimé sur des terminaisons GABAergiques peut avoir des effets de circuit différents de ceux du même récepteur sur des terminaisons glutamatergiques. Un récepteur surexprimé pendant l’inflammation n’est pas équivalent à son état basal. La distribution est dynamique.
Signalisation désigne les conséquences intracellulaires de l’engagement du récepteur. Pour CB1 et CB2, cela comprend le couplage aux protéines G de la famille Gi/o, l’inhibition de l’adénylate cyclase, la modulation des canaux ioniques, des modifications des cascades de kinases, le recrutement de β-arrestin, la désensibilisation du récepteur et son internalisation. Cela inclut aussi la modulation allostérique et l’agonisme biaisé, où les ligands peuvent favoriser certaines issues de signalisation au détriment d’autres. Ce n’est pas un détail académique. C’est souvent la différence entre analgésie, sédation, tolérance, dysphorie ou échec d’un essai.
Cibles médicamenteuses désigne des récepteurs envisagés pour une intervention, et non des succès garantis. Une cible sélective de CB1 peut réduire certains effets hors cible, tout en se heurtant malgré tout à des effets indésirables centraux. Une cible sélective de CB2 peut éviter certains effets intoxicants, mais la sélectivité ne garantit pas l’efficacité dans des maladies humaines complexes. Les travaux de biologie des systèmes le montrent clairement. Une analyse intégrative de réseau indexée dans PubMed en 2025/2026 a identifié CB1 et CB2 comme des nœuds hautement influents du système endocannabinoid et a relié leur signalisation à des voies métaboliques plus larges. Autrement dit, ces récepteurs s’inscrivent dans des réseaux plus vastes. Si l’on agit sur un nœud, d’autres voies se modifient.
Telle est la position de cet article. CB1 et CB2 sont des nœuds de signalisation dépendants du contexte. Pas des interrupteurs statiques. Pas de simples étiquettes pour le « cerveau » et le « système immunitaire ». Si la science du cannabis veut expliquer pourquoi un composé paraît thérapeutique dans un contexte, intoxicant dans un autre, et décevant dans un essai clinique, elle doit commencer au niveau du récepteur et y rester assez longtemps pour suivre la biologie là où elle se déroule réellement.
Une brève histoire de la découverte des récepteurs cannabinoïdes
Avant l’identification des récepteurs cannabinoïdes, la science du cannabis était surtout une histoire de chimie. Les chercheurs pouvaient isoler des composés végétaux, comparer des effets comportementaux bruts chez l’animal et débattre de la puissance, mais ils ne pouvaient pas encore expliquer comment une molécule comme le delta-9-tetrahydrocannabinol, ou THC, produisait ses effets avec une précision comparable à celle d’un niveau récepteur. Cela a changé à la fin des années 1980 et au début des années 1990. Cette évolution a été décisive : la recherche sur le cannabis est passée du catalogage des phytocannabinoids à l’étude des interactions ligand-récepteur, de la signalisation intracellulaire, de la distribution tissulaire et, finalement, du système lipidique endogène désormais appelé système endocannabinoïde, ou ECS.
| Année | Étape importante | Personnes citées dans l’article |
|---|---|---|
| 1964 | Isolement et structure du THC | Raphael Mechoulam ; Yechiel Gaoni |
| 1988 | Sites de liaison cannabinoïdes spécifiques à haute affinité dans les membranes cérébrales de rat | Allyn Howlett ; William Devane |
| 1990 | Clonage de CB1 | Lisa Matsuda et collaborateurs |
| 1992 | Identification de l’anandamide | William Devane ; Lumír Hanuš ; Raphael Mechoulam ; collaborateurs |
| 1993 | Identification de CB2 | Sean Munro ; Kerrie Thomas ; M. Abu-Shaar |
| 1995 | Identification du 2-AG par des groupes distincts | Équipe de Mechoulam ; groupe de Sugiura |
De la pharmacologie du THC à l’identification des récepteurs
Une étape précoce déterminante a eu lieu en 1964, lorsque Raphael Mechoulam et Yechiel Gaoni ont rapporté l’isolement et la structure du THC. Cette avancée était importante, car elle donnait aux pharmacologues une molécule définie à tester plutôt qu’un extrait botanique variable. Au cours des deux décennies suivantes, le domaine a établi une carte structure-activité à partir du THC et de cannabinoid apparentés, mais le mécanisme restait débattu. Certains chercheurs privilégiaient des effets membranaires non spécifiques, les cannabinoid étant lipophiles. Cette hypothèse est devenue plus difficile à défendre à mesure que des données de liaison stéréosélective et saturable s’accumulaient.
L’ère des récepteurs a véritablement commencé avec les études de liaison dans les années 1980. En 1988, Allyn Howlett et William Devane ont publié dans Molecular Pharmacology un article fondateur montrant des sites de liaison cannabinoïdes spécifiques et à haute affinité dans des membranes de cerveau de rat, en utilisant l’agoniste synthétique CP55,940. Le résultat n’était pas une simple suggestion vague d’une cible. Il montrait une saturabilité, une variation régionale et une spécificité pharmacologique compatibles avec un véritable récepteur. Le tissu cérébral ne répondait pas aux cannabinoid comme s’ils se dissolvaient simplement dans les bicouches lipidiques en perturbant tout à la fois. Il existait une sélectivité.
Trois ans plus tard, en 1990, Lisa Matsuda et ses collègues ont cloné le premier récepteur cannabinoïde, désormais appelé CB1, et l’ont publié dans Nature. CB1 a été identifié comme un récepteur couplé aux protéines G, ou GPCR, une découverte qui a immédiatement inscrit la pharmacologie des cannabinoid dans l’une des plus importantes superfamilles de signalisation en biologie. C’était important, car les GPCR ne sont pas de simples interrupteurs. Ils adoptent plusieurs états conformationnels, se couplent à différents partenaires intracellulaires, se désensibilisent, s’internalisent et présentent un biais de signalisation dépendant du ligand. Ces idées deviendraient centrales bien plus tard, mais le clonage de CB1 les a rendues possibles.
CB2 a suivi rapidement. En 1993, Sean Munro, Kerrie Thomas et M. Abu-Shaar ont identifié un second récepteur cannabinoïde, CB2, également dans Nature, initialement caractérisé à partir de tissus liés au système immunitaire. Cette découverte a créé un raccourci durable qui a façonné le domaine pendant des années : CB1 comme « récepteur cérébral » associé à l’intoxication, CB2 comme récepteur « périphérique » ou immunitaire associé à l’inflammation. Ce raccourci était utile, mais déjà trop simpliste, et il a mal vieilli. La distribution des deux récepteurs dépend de l’espèce, du type cellulaire, de l’état d’activation, du contexte pathologique et de la méthode de dosage.
Comment CB1 et CB2 ont transformé le champ de l’endocannabinoïde
Une fois CB1 et CB2 identifiés, la question suivante était évidente : pourquoi l’organisme possédait-il des récepteurs pour des cannabinoid d’origine végétale ? La réponse est arrivée en 1992, lorsque William Devane, Lumír Hanuš, Raphael Mechoulam et leurs collègues ont identifié l’anandamide, formellement l’éthanolamide d’arachidonoyle, comme ligand endogène. L’article, publié dans Science, a marqué une rupture conceptuelle. La pharmacologie du cannabis ne concernait plus seulement des composés exogènes issus de Cannabis sativa. Elle concernait un système de signalisation lipidique natif.
Un second ligand endogène majeur, le 2-arachidonoylglycerol ou 2-AG, a été identifié en 1995 par des groupes distincts, dont l’équipe de Mechoulam et le groupe de Sugiura. Avec les récepteurs et les ligands endogènes en place, l’ECS s’est rapidement développé. Les chercheurs ont identifié des enzymes de synthèse et de dégradation telles que la fatty acid amide hydrolase, FAAH, pour l’anandamide, et la monoacylglycerol lipase, MAGL, pour le 2-AG. Ils ont également été confrontés à une question toujours non résolue : comment ces molécules très lipophiles se déplacent à travers les membranes et l’espace extracellulaire. Le domaine parle souvent de « transport », mais un transporteur endocannabinoïde unique et dédié est resté insaisissable.
C’est à ce moment-là que la science des cannabinoid a cessé d’être un tableau à deux récepteurs pour devenir un réseau de signalisation. CB1 et CB2 ont été reliés aux protéines Gi/o, à l’inhibition de l’adénylate cyclase, à la modulation des canaux calciques et potassiques, et à la suppression de la libération des neurotransmetteurs. Mais l’histoire ne s’est pas arrêtée là. Les récepteurs pouvaient recruter des beta-arrestins, subir une désensibilisation et une internalisation, et répondre différemment aux phytocannabinoids, aux endocannabinoids et aux ligands synthétiques, même lorsque ces ligands ciblaient nominalement le même récepteur. Le langage actuel des GPCR, celui de l’agonisme biaisé, s’applique particulièrement bien aux cannabinoid. Un article de 2025 publié dans American Journal of Psychiatry a soutenu que la signalisation biaisée de CB1 constitue une stratégie thérapeutique plausible pour la schizophrénie, un trouble qui touche environ 24 millions de personnes dans le monde selon l’WHO. C’est bien loin de l’ancienne image de CB1 comme simple récepteur expliquant l’intoxication provoquée par le THC.
L’histoire de CB2 s’est également élargie. Les travaux initiaux le situaient surtout dans les tissus immunitaires, ce qui était globalement juste, mais des études ultérieures ont trouvé une expression de CB2 dans la microglie et, dans certaines conditions, dans d’autres populations cellulaires du système nerveux central. Une revue de 2026 dans Frontiers in Behavioral Neuroscience a décrit « une mise à jour sur les 3 dernières années » reliant la signalisation de CB2 à des mécanismes neuroinflammatoires et neurodégénératifs, montrant clairement que CB2 ne peut pas être considéré comme sans pertinence pour le cerveau. Les travaux structuraux récents sont allés encore plus loin. Une revue de 2026 dans Frontiers in Chemical Biology sur « CB1 and CB2 » a souligné que la sélectivité de sous-type dépend de différences structurales au niveau du récepteur qui modifient la liaison, l’efficacité et la régulation. Une étude récente indexée dans PubMed sur la sélectivité de sous-type soutient de même que la sélectivité des endocannabinoids est dynamique, façonnée par le comportement conformationnel plutôt que par un simple modèle clé-serrure.
Les chercheurs fondateurs et pourquoi cette histoire compte encore
Trois noms doivent figurer au centre de cette histoire. Raphael Mechoulam a contribué à définir la base chimique et biologique de la science des cannabinoid, depuis les travaux sur la structure du THC jusqu’à la découverte des endocannabinoids. Lumír Hanuš a joué un rôle central dans l’identification de l’anandamide et dans les recherches ultérieures sur les endocannabinoids. La pharmacologie des récepteurs menée par Allyn Howlett a été décisive pour démontrer que les cannabinoid agissent par des sites de liaison et des mécanismes de signalisation spécifiques dans le cerveau. Sans leurs travaux, il n’existerait pas de champ moderne de l’ECS.
Cette histoire reste importante, car les anciennes simplifications continuent de fausser les débats actuels. En 2019, on estimait à 200 millions le nombre de personnes dans le monde, soit environ 4 % des personnes âgées de 15 à 64 ans, ayant consommé du cannabis selon l’WHO. Parallèlement, la FDA indique avoir approuvé un produit médicamenteux dérivé du cannabis et trois produits médicamenteux liés au cannabis. L’exposition du public est immense. La transposition clinique est sélective et difficile. L’histoire des récepteurs explique pourquoi. Les effets des cannabinoid dépendent de la classe de ligand, de l’état du récepteur, de la localisation tissulaire, du moment d’exposition et du biais de voie. Ils dépendent aussi d’un réseau plus large. Une analyse intégrative de réseau de 2025/2026 a identifié CB1 et CB2 comme des nœuds hautement influents connectés à des voies métaboliques plutôt que comme des cibles isolées.
C’est là le véritable héritage de la découverte des récepteurs. Elle n’a pas simplifié la biologie du cannabis. Elle a montré pourquoi cette biologie est plus complexe que ne l’autorisait l’ancienne séparation entre cerveau et corps.
Où se trouve CB1 : circuits cérébraux, tissus périphériques et gradients fonctionnels
CB1 a gagné sa réputation de principal récepteur cannabinoïde psychoactif pour une bonne raison. Il est abondant dans le système nerveux central, et les travaux de pharmacologie des récepteurs menés par Allyn Howlett ont contribué à établir que le THC agit par un système récepteur spécifique et saturable plutôt que par des effets membranaires non spécifiques. Mais l’ancien raccourci — CB1 dans le cerveau, CB2 dans les cellules immunitaires — crée désormais plus de confusion que de clarté. CB1 est fortement enrichi dans les circuits neuronaux, certes. Il est aussi présent dans l’intestin, le foie, le tissu adipeux, les organes reproducteurs, les tissus cardiovasculaires et les voies sensorielles, où il module l’alimentation, le métabolisme, la signalisation de la douleur et la fonction autonome. La distribution est large. La fonction est conditionnelle.
Cela compte parce que l’exposition aux cannabinoid est fréquente. L’Organisation mondiale de la Santé a estimé que 200 millions de personnes ont consommé du cannabis en 2019, soit environ 4 % de la population mondiale âgée de 15 à 64 ans. Cela compte aussi parce que la pharmacologie des récepteurs continue de déboucher sur la médecine : la FDA américaine indique qu’un produit médicamenteux dérivé du cannabis et trois produits médicamenteux liés au cannabis sont approuvés. Un récepteur présent dans autant d’organes ne peut pas être réduit à une seule étiquette comportementale.
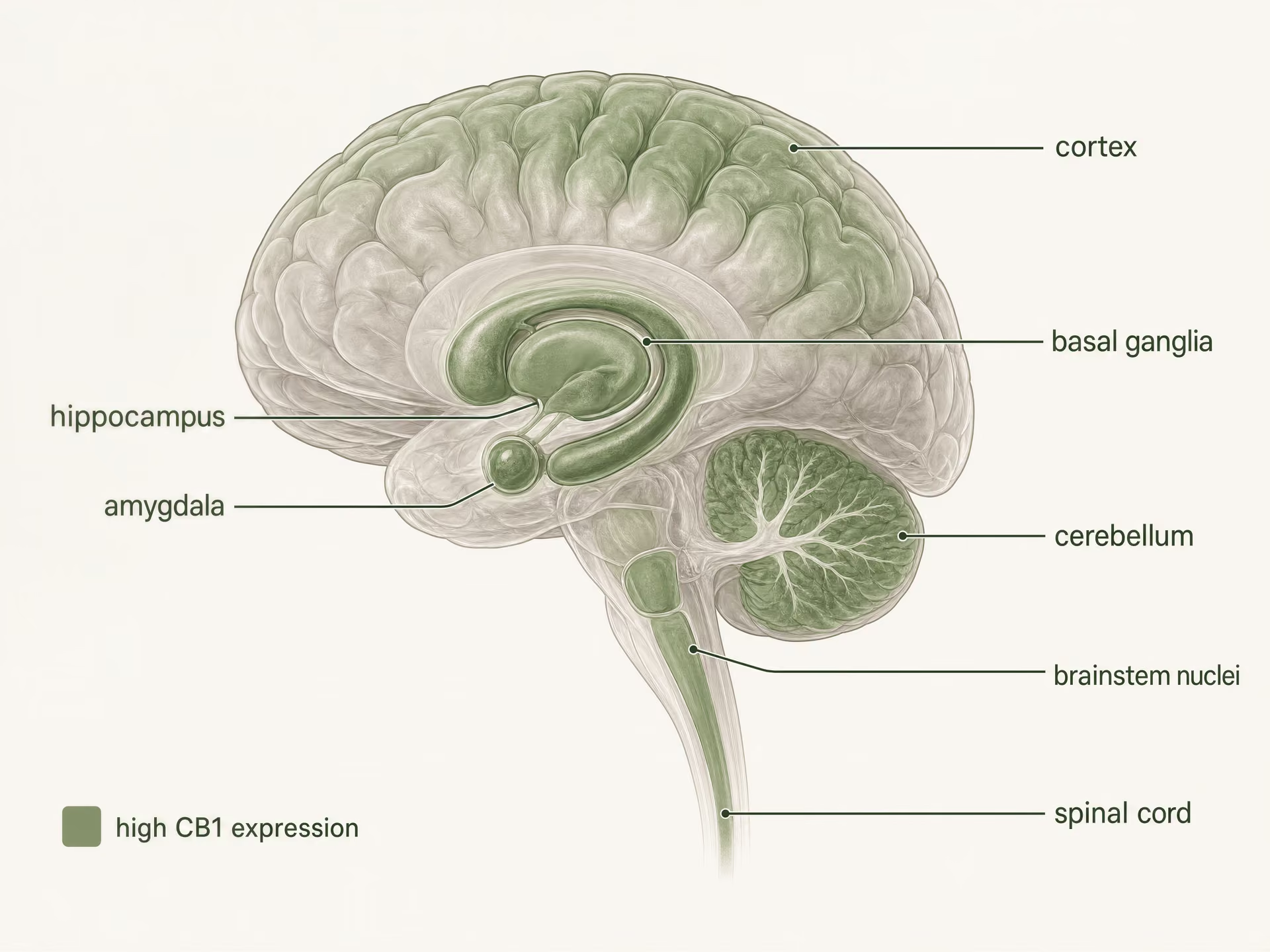
- Schéma général
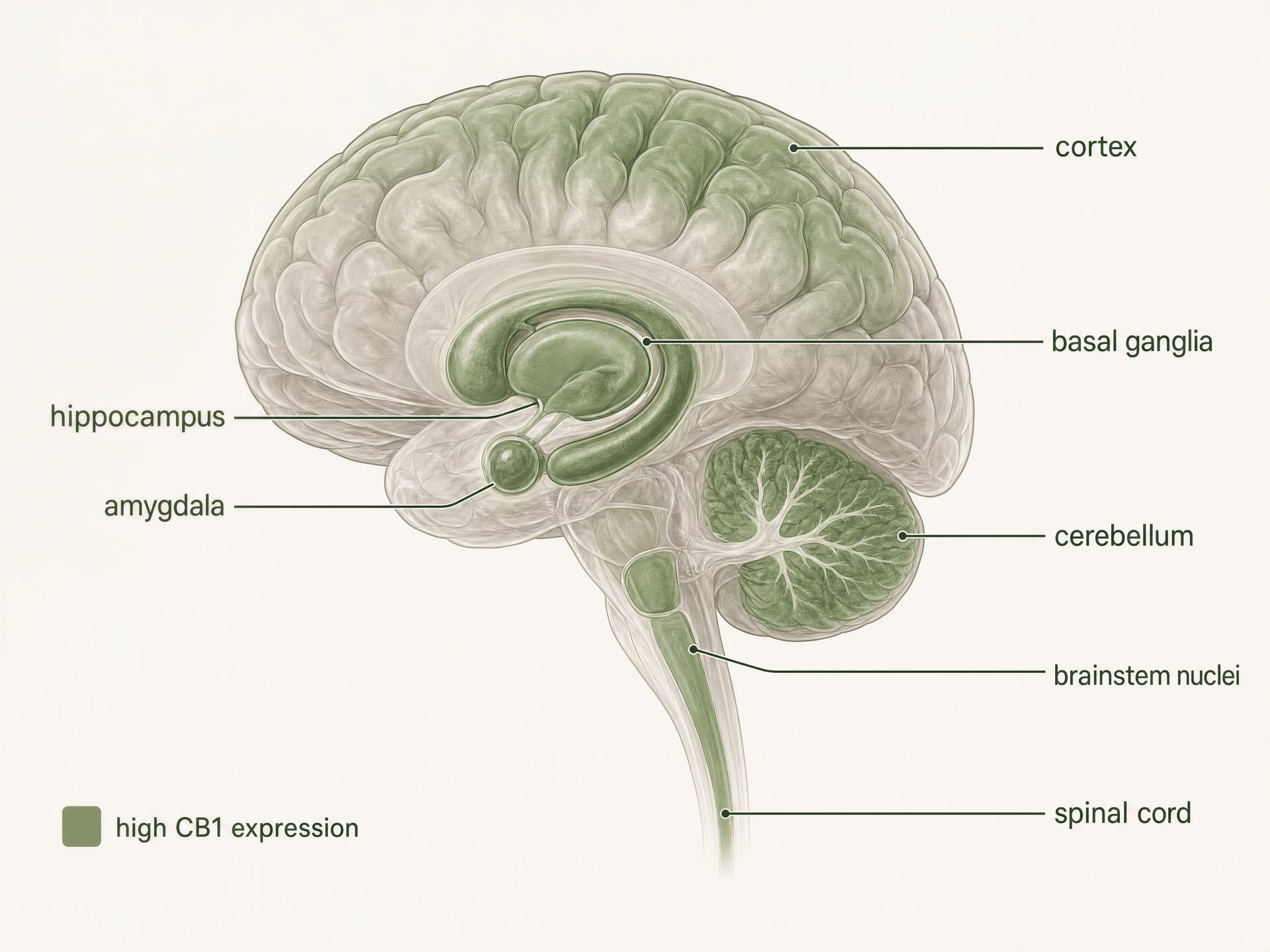
- L’un des GPCR les plus abondants dans le cerveau des mammifères
- Régions à forte densité citées
- Cortex, hippocampe, amygdale, ganglions de la base, cervelet
- Sites liés à la douleur cités
- Substance grise périaqueducale, médulla ventromédiane rostrale, corne dorsale
- Sites périphériques cités
- Intestin, foie, tissu adipeux, voies reproductives, cardiovasculaires et sensorielles
Forte expression dans le système nerveux central
{kind=link}
CB1 est l’un des récepteurs couplés aux protéines G les plus abondants du cerveau des mammifères. L’autoradiographie, l’hybridation in situ et la cartographie immunohistochimique ont établi un tableau clair bien avant les études structurales actuelles : des densités élevées apparaissent dans le cortex, l’hippocampe, l’amygdale, les ganglions de la base, le cervelet et plusieurs régions impliquées dans le traitement de la douleur, avec une expression supplémentaire dans les noyaux du tronc cérébral et tout au long de la moelle épinière. Ce profil correspond de façon étonnamment fidèle, mais pas parfaite, aux effets classiques du THC.
| Localisation de CB1 | Effet synaptique immédiat | Conséquence exemple mentionnée dans l’article |
|---|---|---|
| Terminaisons d’interneurones GABAergiques | Supprime la libération de GABA | Désinhibition des neurones en aval |
| Terminaisons glutamatergiques | Supprime la libération de glutamate | Diminution de l’excitation |
| Circuits des ganglions de la base et du cervelet | Modifie la libération des neurotransmetteurs dans les voies motrices | Ralentissement moteur, modification des circuits d’habitudes, coordination altérée |
| Voies de la douleur | Module la transmission nociceptive | Modifications du traitement ascendant, descendant, inflammatoire et affectif de la douleur |
Dans le cortex et l’hippocampe, CB1 s’inscrit dans des circuits qui régulent l’attention, la mémoire de travail, l’apprentissage de l’extinction et la plasticité synaptique à court terme. Les effets sur la mémoire ne se résument pas à « hippocampe égale oubli ». Ils dépendent fortement des terminaisons axonales qui expriment le récepteur. CB1 est souvent concentré de manière présynaptique, où il inhibe la libération de neurotransmetteurs après activation par des endocannabinoid tels que l’anandamide et le 2-arachidonoylglycérol, des lipides de signalisation dont la découverte par Raphael Mechoulam, Lumír Hanuš et leurs collègues a transformé le domaine. Lorsque CB1 est activé sur des terminaisons d’interneurones GABAergiques, il peut lever l’inhibition des neurones en aval ; lorsqu’il est activé sur des terminaisons glutamatergiques, il peut atténuer l’excitation. Même récepteur, résultat réseau opposé.
Les ganglions de la base et le cervelet expliquent un autre ensemble d’effets bien connus. L’expression dense de CB1 dans le striatum, le globus pallidus, la substance noire pars reticulata et les couches moléculaires du cervelet relie l’activation du récepteur au ralentissement moteur, à la modification des circuits de l’habitude, à l’altération de la coordination et, à certaines doses, à des effets de type catalepsie dans les modèles animaux. Pourtant, le fait que CB1 soit peu exprimé dans les centres bulbaires cardiorespiratoires par rapport aux récepteurs opioïdes aide à expliquer pourquoi un surdosage de cannabinoid ne produit généralement pas le même schéma de dépression respiratoire fatale que celui observé avec de puissants agonistes opioïdes. La localisation compte. L’absence compte aussi.[5]The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research. National Academies of Sciences, Engineering, and Medicine. National Academies Press, 2017. https://nap.nationalacademies.org/catalog/24625/the-health-effects-of-cannabis-and-cannabinoids-the-current-state
Le traitement de la douleur montre la même logique régionale. CB1 se trouve dans la substance grise périaqueducale, la médulla ventromédiane rostrale, la corne dorsale de la moelle épinière et les voies nociceptives périphériques. Cela confère au récepteur plusieurs points d’entrée dans la nociception : il peut modifier les signaux ascendants de la douleur, le contrôle descendant de la douleur, la sensibilisation inflammatoire et la coloration émotionnelle de la douleur. C’est l’une des raisons pour lesquelles les cannabinoid sont restés présents dans le débat sur la douleur chronique, d’autant plus que près d’un adulte sur cinq aux États-Unis vit avec une douleur chronique, selon les National Academies. Mais l’analgésie n’est pas garantie simplement parce que CB1 est présent. La sédation, l’altération cognitive, la tolérance et les effets indésirables limitant la dose apparaissent souvent via des circuits voisins ou via les mêmes circuits à différents niveaux d’engagement du récepteur.
La signalisation biaisée de CB1 peut séparer les effets thérapeutiques recherchés des effets psychoactifs ou cognitifs indésirables.Limited evidence
Protéines Gi/o Famille de protéines G qui réduisent souvent l’activité de l’adénylate cyclase et aident à contrôler les canaux ioniques après activation d’un GPCR.
La biologie moderne des récepteurs ajoute une couche supplémentaire. CB1 n’est pas un simple interrupteur marche/arrêt. Il est principalement couplé aux protéines Gi/o, réduisant l’activité de l’adénylate cyclase et modulant les canaux ioniques, mais il peut aussi recruter les bêta-arrestines, subir une désensibilisation et une internalisation, et présenter un biais de signalisation dépendant du ligand. L’article de 2025 de l’American Journal of Psychiatry soutenant que la signalisation biaisée de CB1 pourrait être exploitée à visée thérapeutique dans la schizophrénie le souligne directement : l’occupation du récepteur est un mauvais prédicteur du résultat. La schizophrénie touchant environ 24 millions de personnes dans le monde, l’idée de séparer les signaux souhaités des effets psychoactifs ou cognitifs indésirables est évidemment attrayante. Reste à savoir si cette séparation est réalisable en pratique : c’est une question ouverte de développement du médicament, et non un fait établi.
CB1 périphérique dans l’intestin, le foie, le tissu adipeux et au-delà
CB1 en dehors du cerveau n’est pas un détail. Il est au cœur de la manière dont les cannabinoid influencent l’appétit, la nausée, la gestion du glucose, le métabolisme lipidique et la sensation viscérale.
Dans l’intestin, CB1 est exprimé dans le système nerveux entérique, les compartiments épithéliaux et les voies liées au nerf vague. Son activation peut ralentir la vidange gastrique, modifier la motilité intestinale, réduire les vomissements et changer la signalisation entre l’intestin et le cerveau. Les effets sur l’appétit sont souvent décrits comme s’ils provenaient uniquement des centres hypothalamiques de la récompense et de l’alimentation, mais CB1 périphérique contribue au phénomène en façonnant les entrées sensorielles et hormonales avant même que les signaux n’atteignent ces circuits. Un repas n’agit pas sur un paysage récepteur vierge ; il modifie localement le tonus des endocannabinoid.
Dans le foie et le tissu adipeux, CB1 participe à la régulation métabolique, notamment la lipogenèse, la sensibilité à l’insuline et le stockage énergétique. C’était l’un des grands enseignements de l’ère du rimonabant. Le blocage de CB1 améliorait le poids et les marqueurs métaboliques, ce qui soutenait l’idée qu’une signalisation endocannabinoid hyperactive contribue à la pathologie liée à l’obésité. Mais le rimonabant, un inverse agoniste de CB1 à action centrale, a aussi provoqué de graves effets indésirables psychiatriques, notamment dépression et anxiété, et a été retiré du marché. Cet épisode est souvent cité comme un échec du « ciblage de CB1 ». Plus exactement, il s’agissait de l’échec d’un type particulier de ciblage de CB1 : un antagonisme central puissant ou une inverse agonisme dans un système récepteur imbriqué dans les circuits de l’humeur et du stress. La leçon n’est pas que CB1 périphérique est sans importance ; la leçon est que le profil d’exposition du médicament et l’état du récepteur comptent autant que le nom du récepteur.
Les adipocytes, les hépatocytes, le tissu pancréatique, le muscle squelettique, les tissus cardiovasculaires et les organes reproducteurs complètent cette cartographie périphérique. Les neurones sensoriels aussi. L’analyse intégrative en réseau indexée dans PubMed en 2025/2026 qui a identifié CB1 et CB2 comme des nœuds hautement influents dans la signalisation des endocannabinoid est utile ici, car elle déplace le cadre d’une simple localisation du récepteur vers la participation du récepteur aux réseaux métaboliques et de signalisation. Un récepteur faiblement exprimé dans un tissu peut néanmoins exercer des effets systémiques majeurs s’il occupe un goulot d’étranglement dans la signalisation locale.
Les travaux structuraux maintiennent également ce débat ancré dans le réel. La revue de 2026 de Frontiers in Chemical Biology sur CB1 et CB2 souligne que la sélectivité et l’efficacité des ligands découlent de différences structurales au niveau du récepteur, qui modifient la liaison, la signalisation et la régulation du récepteur. Une étude indexée dans PubMed en 2025/2026 sur la sélectivité des sous-types soutient de même que la dynamique conformationnelle, et pas seulement l’ajustement de type serrure-clé, façonne la manière dont les endocannabinoid distinguent les sous-types de récepteurs. Cela compte pour la distribution de CB1, car « CB1 dans le foie » ne signifie pas que THC, anandamide, 2-AG et un agoniste synthétique y produiront tous le même effet.
Pourquoi la distribution n’équivaut pas à une fonction uniforme unique
La plus grande erreur dans les cartes des récepteurs est de traiter l’expression comme un destin. Ce n’en est pas un. Une expression élevée indique où chercher, pas ce qui va se produire.
Premièrement, le type cellulaire change le signe de l’effet. Un récepteur CB1 sur une terminaison glutamatergique peut réduire l’excitation. Le même récepteur sur une terminaison GABAergique peut réduire l’inhibition. Ces résultats ne sont pas interchangeables. Deuxièmement, la localisation synaptique compte. CB1 est généralement présynaptique, souvent activé par des endocannabinoid libérés « à la demande » par des neurones postsynaptiques, créant un contrôle rétrograde de la libération des neurotransmetteurs. Cette organisation favorise une modulation brève, dépendante de l’activité, plutôt qu’une activation constante du récepteur.
Troisièmement, l’identité du ligand compte. Les endocannabinoid sont des messagers locaux de courte durée. Les phytocannabinoid tels que THC arrivent de l’extérieur du système, souvent à des expositions plus élevées et plus durables que les signaux endogènes. Les ligands synthétiques peuvent agir encore plus fortement, avec une efficacité et un biais différents. Certains favorisent plus fortement la signalisation Gi/o ; d’autres favorisent le recrutement des bêta-arrestines, la désensibilisation ou l’internalisation du récepteur. C’est pourquoi deux composés peuvent tous deux être appelés agonistes de CB1 tout en différant nettement par la stimulation de l’appétit, l’altération de la mémoire, l’atteinte motrice et le développement de la tolérance.
Quatrièmement, la disponibilité locale du ligand change tout. L’anandamide et le 2-AG sont produits et dégradés sur place, de sorte que leurs effets dépendent de l’activité neuronale, de l’état métabolique, de l’expression enzymatique et du contexte inflammatoire. Cinquièmement, la densité du récepteur elle-même suit un gradient. La région cérébrale, le stade du développement, l’état pathologique et l’exposition répétée au médicament modifient tous les niveaux de CB1 et sa réactivité.
La littérature actuelle s’éloigne des dichotomies précisément pour cette raison. La revue de 2026 de Frontiers in Behavioral Neuroscience note une mise à jour, au cours des trois dernières années, de la compréhension de la signalisation des récepteurs aux cannabinoid dans les troubles du SNC, en particulier lorsque les mécanismes neuroinflammatoires et neurodégénératifs sont inclus. CB1 doit être interprété avec la même prudence. C’est un récepteur central dominant, mais non exclusivement central ; un récepteur de l’alimentation, mais pas seulement ; une cible de la douleur, mais pas un interrupteur analgésique pur. Toute description sérieuse de la distribution de CB1 doit penser en gradients, en circuits et en états de signalisation plutôt qu’en caricature cerveau contre corps.
Où se trouve CB2 : racines immunitaires et cartographie croissante du SNC
L’ancienne formule le disait simplement : CB1 est le récepteur du cerveau, CB2 est le récepteur immunitaire. Cette représentation a aidé l’enseignement initial, mais elle induit désormais davantage en erreur qu’elle n’éclaire. CB2 présente effectivement un enrichissement classique en dehors des neurones, en particulier dans les lignées immunitaires et hématopoïétiques, et ce fait reste important pour la pharmacologie. Pourtant, la littérature récente, notamment la revue 2026 de Frontiers in Behavioral Neuroscience, formule une thèse plus forte : CB2 est désormais discuté dans les troubles du système nerveux central parce que son expression et sa signalisation deviennent plus visibles dans les microglies, les circuits inflammatoires et les états liés aux lésions, une « mise à jour au cours des trois dernières années » qui a changé la manière dont le récepteur est cartographié et interprété. Il ne s’ensuit pas que CB2 soit soudain devenu un récepteur cérébral pan-neuronal abondant. Ce n’est pas le cas. Il s’ensuit que la distribution du récepteur doit être décrite comme conditionnelle, spécifique au type cellulaire et dépendante de l’état.
Cette distinction a une importance clinique. L’Organisation mondiale de la Santé a estimé que 200 millions de personnes ont utilisé cannabis en 2019, soit 4 % de la population mondiale âgée de 15 à 64 ans. Même avec seulement un petit nombre de médicaments liés au cannabinoid approuvés — la FDA en 2025 en comptait un produit dérivé du cannabis et trois produits liés au cannabis — la localisation des récepteurs façonne toujours les zones dans lesquelles les développeurs de médicaments recherchent des effets anti-inflammatoires, analgésiques, neuroprotecteurs et psychiatriques, ainsi que les domaines où ils anticipent des effets indésirables.
- Enrichissement classique
- Cellules immunitaires et hématopoïétiques
- Types cellulaires cités
- Lymphocytes B, lymphocytes T, macrophages, monocytes, cellules tueuses naturelles, neutrophiles, mastocytes
- Tissus canoniques cités
- Rate, amygdale, thymus, moelle osseuse, cellules immunitaires circulantes
- Pertinence pour le SNC soulignée
- Microglie et états liés à la pathologie
Enrichissement classique dans les cellules immunitaires et hématopoïétiques
CB2 a été initialement identifié comme le sous-type de récepteur cannabinoid présentant l’expression la plus forte dans les cellules liées à l’immunité plutôt qu’à la transmission synaptique rapide. C’est toujours le bon point de départ. Par rapport à CB1, qui est largement représenté dans de nombreuses populations neuronales, CB2 est classiquement enrichi dans les cellules B, les cellules T, les macrophages, les monocytes, les cellules natural killer, les neutrophiles, les mast cells et d’autres compartiments hématopoïétiques. La rate, l’amygdale, le thymus, la moelle osseuse et les populations circulantes de cellules immunitaires ont donc été les tissus canoniques pour l’analyse de CB2.
Cette orientation immunitaire a façonné l’idée initiale de développement pharmacologique d’agonistes sélectifs de CB2, afin de capter des bénéfices anti-inflammatoires ou analgésiques tout en évitant les effets intoxicants associés à une forte activation de CB1 dans le cerveau. C’était une hypothèse sensée, mais incomplète. CB2 est un GPCR couplé à Gi/o, et comme CB1, il ne se contente pas de s’activer ou de se désactiver. Selon le ligand, la conformation du récepteur et le contexte cellulaire, CB2 peut réduire l’activité de l’adénylate cyclase, influencer les voies MAPK, modifier indirectement le couplage des canaux ioniques, recruter les beta-arrestins, et subir une désensibilisation ou une internalisation. Ainsi, même dans les tissus immunitaires périphériques, la vraie question n’est pas seulement de savoir si CB2 est présent, mais quelles cellules l’expriment, à quel niveau, sous quel stimulus et avec quel biais en aval.
Cette complexité explique en partie pourquoi des ligands apparemment similaires peuvent se comporter différemment. La revue 2026 de Frontiers in Chemical Biology sur la structure des récepteurs cannabinoid soutient que la sélectivité vis-à-vis de « CB1 et CB2 » est façonnée par des différences structurelles au niveau du récepteur, qui modifient la liaison du ligand, l’efficacité de signalisation et la régulation du récepteur. Une étude indexée PubMed de 2025/2026 sur la sélectivité des sous-types a poussé l’idée plus loin en montrant que la sélectivité des endocannabinoid est dynamique et liée à des états conformationnels, et non à un modèle rigide de clé et serrure. Cela importe pour la cartographie tissulaire, car un ligand endogène tel que le 2-AG ou l’anandamide, un phytocannabinoid tel que THC, et un agoniste synthétique préférant CB2 peuvent tous rencontrer la même population de récepteurs, mais stabiliser des résultats de signalisation différents.
L’ancienne carte de CB2 centrée sur l’immunité n’était donc pas fausse. Elle était incomplète. CB2 reste mieux décrit comme un récepteur ayant de fortes racines dans le système immunitaire. Mais les racines ne constituent pas tout l’organisme.
| Contexte | Comment CB2 est décrit | Point d’interprétation |
|---|---|---|
| Cerveau sain de référence | Souvent faible ou proche des limites de détection dans de nombreuses régions | Un faible signal basal ne signifie pas absence de pertinence |
| Microglie activée | Plus détectable après lésion ou inflammation | Soutient la pertinence pour le SNC par des fonctions de type immunitaire |
| Astrocytes / endothéliales / cellules infiltrantes | Signalé dans certains contextes pathologiques | La localisation dépend de la méthode et du modèle |
| Expression neuronale constitutive étendue | Nécessite des preuves plus solides | L’article traite cette affirmation avec prudence |
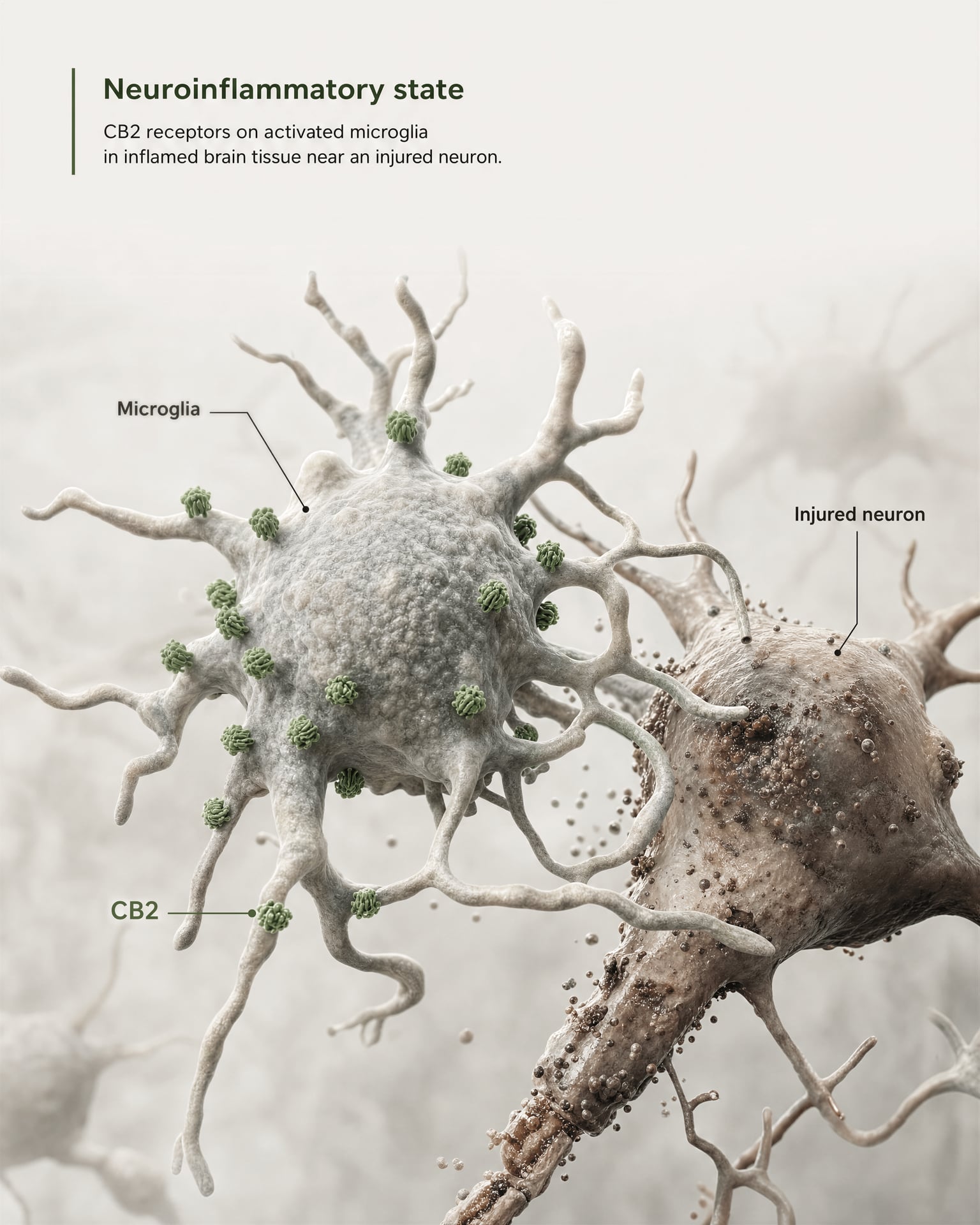
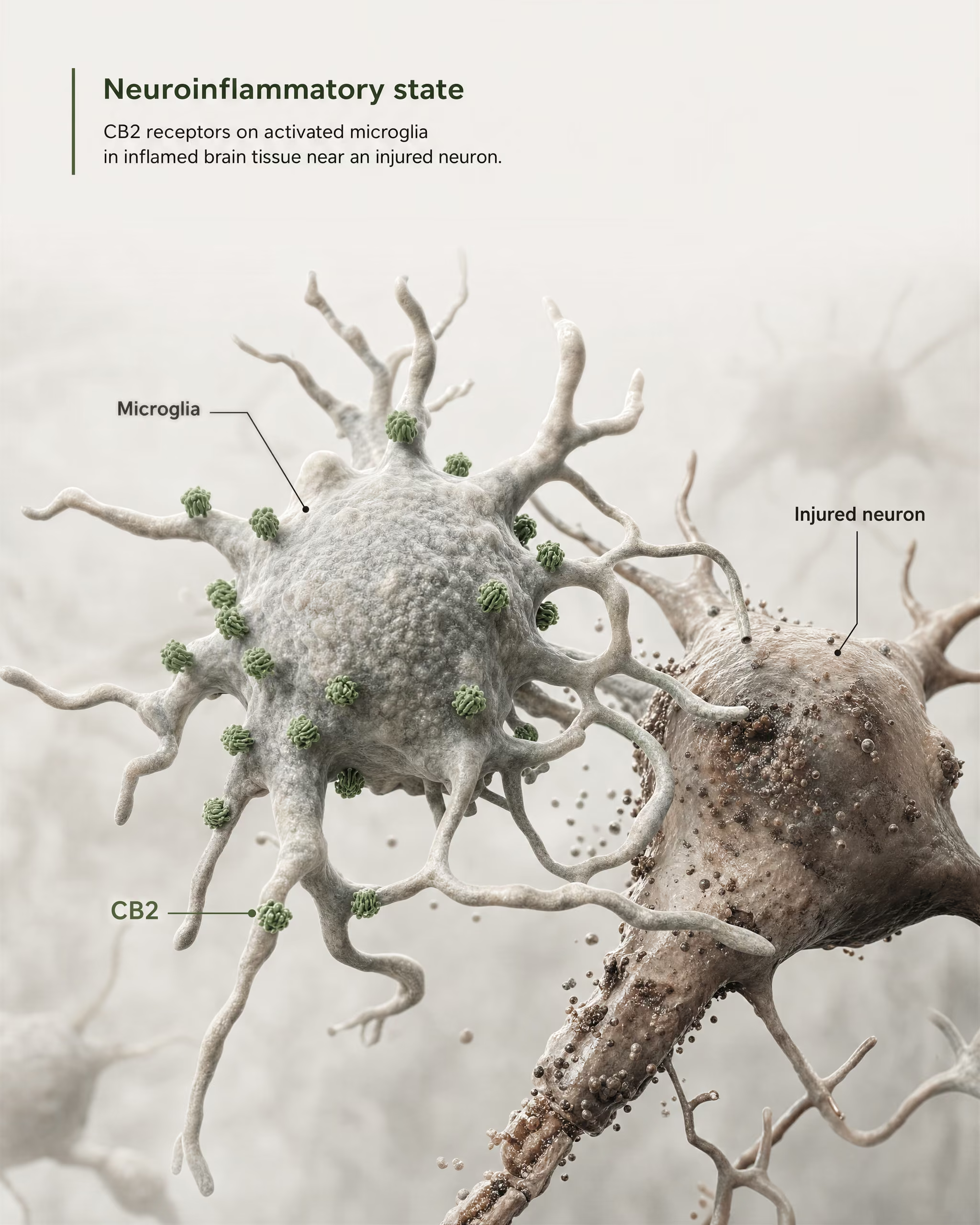
CB2 dans la microglie, la neuroinflammation et les états de lésion
L’argument le plus solide en faveur d’une pertinence centrale ne consiste pas à affirmer que CB2 est largement abondant sur les neurones sains du prosencéphale. Il repose sur la microglie et sur la biologie des maladies.
{kind=link}
La microglie est la cellule immunitaire résidente du SNC, et elle se situe précisément à la frontière où l’ancien modèle de « récepteur immunitaire périphérique » commence à échouer. Si un récepteur est exprimé dans le système de surveillance immunitaire et de réponse inflammatoire propre au cerveau, alors le qualifier de simplement périphérique devient inexact. La revue 2026 de Frontiers in Behavioral Neuroscience formule directement ce point : la signalisation de CB2 a attiré l’attention dans les troubles du SNC parce qu’elle est liée à des mécanismes neuroinflammatoires et neurodégénératifs. C’est pourquoi CB2 apparaît désormais dans les discussions sur la maladie d’Alzheimer, la maladie de Parkinson, la sclérose en plaques, les traumatismes crâniens, l’AVC, la douleur neuropathique et certaines affections psychiatriques où la signalisation inflammatoire fait partie de la pathologie.
L’expression clé est l’expression induite ou augmentée. Dans de nombreuses régions cérébrales saines, l’expression basale de CB2 est faible, parfois proche des limites des anciennes méthodes de détection. Mais après une lésion, une infection, une inflammation chronique ou une neurodégénérescence, le signal de CB2 devient souvent plus détectable, en particulier dans la microglie activée et, dans certaines études, dans les astrocytes, les cellules immunitaires infiltrantes, les compartiments endothéliaux ou des sous-ensembles neuronaux restreints. Il s’agit d’une règle de distribution très différente de celle généralement appliquée à CB1. CB1 est souvent abondant de manière constitutive dans des circuits neuronaux définis. CB2 est plus souvent interprété comme un récepteur dont la pertinence dans le SNC émerge sous l’effet du stress, de la pathologie ou de l’activation inflammatoire.
Cette distinction a des conséquences pratiques. Un médicament ciblant CB2 peut avoir peu d’effet dans un tissu sain où la densité du récepteur est faible, mais montrer une activité mesurable dans un tissu malade où l’expression a augmenté et où les réseaux de signalisation ont changé. Cette inducibilité explique en partie pourquoi les résultats précliniques ont été à la fois enthousiasmants et difficiles à transposer. Le moment compte. Le stade de la maladie compte. La composition cellulaire compte. Un environnement microglial post-lésionnel n’est pas pharmacologiquement équivalent à une tranche cérébrale non stimulée.
Les problèmes d’interprétation ne sont pas triviaux. CB2 a une longue histoire de préoccupations concernant la spécificité des anticorps, de détection de transcrits à faible abondance, de différences entre espèces et d’affirmations de localisation incohérentes selon les méthodes. Certains rapports précoces ont probablement surestimé CB2 neuronal en raison d’outils insuffisants. C’est pourquoi les études rigoureuses s’appuient désormais sur des preuves convergentes — transcriptomique monocellulaire, hybridation in situ, rapporteurs génétiques validés, contrôles knockout, données protéomiques lorsque cela est possible, et comparaisons dépendantes de l’état — plutôt que sur un seul résultat de marquage. Si une étude rapporte CB2 dans les neurones à l’état basal et qu’une autre ne le détecte pas, l’écart peut refléter de vraies différences régionales, le statut de la maladie, l’espèce, l’âge, ou simplement les limites du test.
CB2 a une pertinence significative pour le SNC, surtout dans les contextes gliaux et inflammatoires liés à une lésion.Limited evidence
La meilleure position actuelle est donc prudente mais claire : CB2 a une réelle pertinence dans le SNC, principalement par des fonctions gliales et de type immunitaire, et cette pertinence augmente pendant la neuroinflammation et les lésions. Les affirmations d’une expression neuronale constitutive et généralisée de CB2 dans le cerveau normal exigent davantage de preuves que les affirmations d’une présence de CB2 dans la microglie et dans les contextes pathologiques.
Comment les 3 dernières années ont modifié le débat sur CB2
La revue 2026 de Frontiers in Behavioral Neuroscience présente explicitement la littérature récente comme une « mise à jour sur les 3 dernières années », et cette formulation traduit un véritable changement. Le débat est passé de la question de savoir si CB2 est « présent dans le cerveau ou non » à celle de savoir où, quand et dans quels états pathologiques sa signalisation devient exploitable.
Trois évolutions ont conduit à ce changement. Premièrement, les méthodes à résolution cellulaire se sont améliorées. Les ensembles de données RNA monocellulaires et mononucléaires, une meilleure cartographie spatiale et des normes de validation plus strictes ont réduit le risque qu’une expression faible ou induisible soit écartée simplement parce que les anciens tests manquaient de sensibilité. Deuxièmement, la neuroinflammation est devenue centrale dans de nombreux modèles de maladies cérébrales. Une fois les maladies analysées à travers des mécanismes immunitaires et gliaux plutôt que dans des cadres limités aux neurones, CB2 est devenu beaucoup plus difficile à ignorer. Troisièmement, la pharmacologie des récepteurs s’est elle-même affinée. Le domaine pense désormais davantage en termes d’efficacité, de biais de signalisation, de trafic des récepteurs et de réponses dépendantes du contexte qu’en termes simples d’occupation.
Cette perspective GPCR plus large est visible même en dehors de la littérature sur CB2. L’article de 2025 de American Journal of Psychiatry sur la signalisation biaisée de CB1 et la schizophrénie soutient que la pharmacologie cannabinoid doit être comprise à travers la signalisation biaisée plutôt que par une simple activation brute des récepteurs. La schizophrénie touche environ 24 millions de personnes dans le monde, selon WHO, ce qui en fait loin d’être une question académique secondaire. La même logique s’applique à CB2. Un ligand « sélectif de CB2 » sur le papier peut néanmoins produire des effets différents selon qu’il active préférentiellement la signalisation par protéine G, le recrutement de beta-arrestins, l’internalisation du récepteur ou des programmes transcriptionnels anti-inflammatoires dans la microglie activée.
La vision systémique plus récente renforce cela. Une étude d’analyse de réseau indexée PubMed de 2025/2026 a identifié CB1 et CB2 comme des nœuds hautement influents du système endocannabinoid et a relié la signalisation des récepteurs aux voies métaboliques plutôt que d’isoler les récepteurs du reste de la biologie cellulaire. Cela correspond à ce que montrent les données sur CB2 dans le SNC : la distribution n’est pas une entrée fixe d’atlas. Elle fait partie d’un réseau de signalisation adaptatif.
En somme, CB2 doit toujours être présenté comme un récepteur cannabinoid enrichi dans le système immunitaire. Mais s’arrêter là donne désormais une image fausse. Dans le cerveau, CB2 est mieux compris comme un récepteur à faible expression basale, inducible et lié à la maladie, dont l’importance devient la plus claire dans la microglie et les états neuroinflammatoires — et dont la détection dépend encore fortement de la méthode, du modèle et du moment.
Comment les récepteurs cannabinoïdes signalent : couplage Gi/o, seconds messagers et effets synaptiques
La pharmacologie des récepteurs cannabinoïdes commence par une affirmation simple qui se complique très vite : CB1 et CB2 sont des récepteurs couplés aux protéines G de classe A, et tous deux signalent le plus souvent via les protéines Gi/o. Ce fait fondamental, établi par les travaux pionniers sur les récepteurs menés par Allyn Howlett et d’autres, demeure valable. Ce qui a changé, c’est la compréhension de ce que signifie réellement le couplage à Gi/o dans les cellules. Cela ne désigne pas un effet en aval unique. Cela désigne un ensemble d’effets possibles dont la combinaison dépend du ligand, de la densité des récepteurs, de l’état de phosphorylation, de l’environnement membranaire, du type cellulaire et du moment.
Cette distinction est importante car environ 200 millions de personnes ont utilisé cannabis en 2019, soit 4 % de la population mondiale âgée de 15 à 64 ans selon l’Organisation mondiale de la Santé, tandis que la FDA indique qu’un médicament dérivé de cannabis et trois médicaments liés au cannabis sont approuvés en 2025. La signalisation des récepteurs n’est pas un sujet secondaire. C’est le mécanisme qui distingue un médicament antiépileptique utile d’une sédation, un médicament contre l’appétit qui échoue d’effets indésirables psychiatriques, et un ligand sélectif en laboratoire d’un ligand décevant en clinique.
Signalisation GPCR canonique de CB1 et CB2
Séquence canonique de signalisation CB1/CB2
- Liaison du ligand Un agoniste stabilise une conformation active du récepteur.
- Activation des protéines G Le récepteur favorise l’échange GDP-GTP sur Gi/o.
- Séparation des sous-unités Galpha et Gbeta-gamma régulent les effecteurs en aval.
- Modification du second messager L’activité de l’adénylate cyclase diminue et l’AMPc baisse.
- Effet cellulaire Les canaux ioniques, la libération des neurotransmetteurs, les kinases et la régulation génique se modifient.
Dans le schéma canonique, la liaison de l’agoniste stabilise une conformation réceptrice active, le récepteur agit comme facteur d’échange de nucléotide de guanine pour Gi/o, Gαi/o échange le GDP contre le GTP, puis les composants Gα et Gβγ régulent des effecteurs en aval. Pour CB1 et CB2, l’indicateur classique est l’inhibition de l’adénylyl cyclase et la diminution de l’AMPc intracellulaire. Cette observation est devenue l’une des premières signatures biochimiques utilisées pour définir l’activité des récepteurs cannabinoïdes.
Mais « canonique » ne doit pas être interprété comme « uniforme ». CB1 présente une activité constitutive élevée dans plusieurs systèmes d’expression, ce qui signifie que le récepteur peut signaler de manière mesurable même en l’absence d’agoniste. Cette propriété aide à expliquer pourquoi les agonistes inverses comme le rimonabant faisaient davantage que bloquer le tonus endocannabinoïde endogène ; ils abaissaient la signalisation en dessous du niveau basal et provoquaient des effets centraux indésirables marqués. CB2 se couple aussi à Gi/o, mais la manière dont les ligands stabilisent les états actifs diffère de CB1. Des travaux structuraux examinés dans Frontiers in Chemical Biology en 2026 ont souligné que la sélectivité entre les sous-types « CB1 et CB2 » est déterminée par des différences au niveau des récepteurs qui modifient non seulement la liaison, mais aussi l’efficacité et la régulation. Une étude indexée dans PubMed en 2025/2026 sur la sélectivité des sous-types a poussé l’analyse plus loin en soutenant que la sélectivité des endocannabinoids est dynamique, façonnée par le comportement conformationnel plutôt que par un modèle fixe de clé et de serrure.
C’est l’une des raisons pour lesquelles phytocannabinoids, endocannabinoids et ligands synthétiques ne doivent jamais être traités comme interchangeables. L’anandamide, identifiée par Raphael Mechoulam et Lumír Hanuš, ainsi que le 2-arachidonoylglycérol, sont des ligands endogènes produits à la demande et rapidement inactivés. Δ9-THC est un agoniste partiel d’origine végétale dont la cinétique et l’efficacité diffèrent de celles de ces endocannabinoids. Des agonistes synthétiques tels que CP55,940, WIN55,212-2 ou HU-210 induisent souvent une activation plus forte du récepteur et peuvent recruter les voies de signalisation à des degrés différents. Certains ligands favorisent la signalisation protéine G au détriment du recrutement de β-arrestine ; d’autres non. L’article du American Journal of Psychiatry en 2025 l’a formulé directement pour CB1, soutenant que la signalisation biaisée est une stratégie thérapeutique plausible dans la schizophrénie, un trouble touchant environ 24 millions de personnes dans le monde.
CB2 apporte une correction supplémentaire aux simplifications anciennes. Il reste enrichi dans de nombreuses populations de cellules immunitaires, mais la revue de 2026 dans Frontiers in Behavioral Neuroscience a décrit « une mise à jour sur les 3 dernières années » au cours de laquelle la signalisation de CB2 a gagné en importance dans des troubles du système nerveux central liés à la neuroinflammation et à la neurodégénérescence. Ainsi, même avant d’aborder l’agonisme biaisé, l’ancienne opposition binaire entre un « récepteur cérébral » et un « récepteur immunitaire » échoue déjà au niveau du contexte de signalisation.
Effets sur l’AMPc, les canaux ioniques et la libération de neurotransmetteurs
L’effet de second messager le mieux connu pour les deux récepteurs est la suppression de la formation d’AMPc par inhibition de l’adénylyl cyclase. Une baisse de l’AMPc signifie souvent une diminution de l’activité de la protéine kinase A, une phosphorylation modifiée des cibles en aval et des changements plus lents dans l’expression génique via des voies telles que CREB. Dans les neurones, toutefois, les effets rapides sont souvent plus importants que les effets lents.
CB1 est fortement positionné pour un contrôle présynaptique. Dans de nombreux circuits cérébraux, il se trouve sur les terminaisons axonales, où l’activation du récepteur réduit la probabilité de libération des neurotransmetteurs. Cela se produit par une combinaison d’inhibition, médiée par Gβγ, des canaux calciques voltage-dépendants et d’activation de conductances potassiques rectifiantes entrantes ou d’autres courants potassiques qui diminuent l’excitabilité de la terminaison. Moins d’entrée de calcium signifie moins de fusion de vésicules. Il en résulte une moindre libération de neurotransmetteur dans la fente synaptique.
C’est le mécanisme central de la signalisation endocannabinoïde rétrograde à courte portée. Un neurone postsynaptique devient actif, synthétise à la demande des endocannabinoids à partir de précurseurs lipidiques membranaires, puis les envoie en sens inverse à travers la synapse pour activer les récepteurs CB1 présynaptiques. La terminaison présynaptique libère alors moins de neurotransmetteur. Il s’agit d’un frein de rétroaction. Dans les synapses excitatrices, cela peut supprimer la libération de glutamate ; dans les synapses inhibitrices, cela peut supprimer la libération de GABA. La direction de la sortie du circuit dépend de la terminaison qui exprime CB1. Même récepteur, conséquence réseau opposée.
| Terme | Ce qui est supprimé | Mécanisme décrit |
|---|---|---|
| DSI | Inhibition | L’activité postsynaptique libère des endocannabinoïdes qui activent CB1 présynaptique et réduisent la libération de GABA |
| DSE | Excitation | L’activité postsynaptique libère des endocannabinoïdes qui activent CB1 présynaptique et réduisent la libération de glutamate |
DSI et DSE Formes à court terme de plasticité synaptique médiée par les endocannabinoïdes, dans lesquelles la dépolarisation postsynaptique supprime la transmission inhibitrice (DSI) ou excitatrice (DSE) par activation présynaptique de CB1.
Les termes physiologiques classiques le décrivent bien : suppression de l’inhibition induite par la dépolarisation, DSI, et suppression de l’excitation induite par la dépolarisation, DSE. Ces deux phénomènes sont des formes de plasticité synaptique à court terme provoquées par la libération d’endocannabinoids et l’activation présynaptique de CB1. Des effets plus durables existent aussi, notamment une dépression à long terme médiée par les endocannabinoids dans certaines synapses. Ces phénomènes sont importants parce qu’ils relient la biochimie des récepteurs au comportement : traitement de la douleur, extinction de la peur, apprentissage des habitudes, appétit, contrôle moteur et seuil convulsif dépendent tous de ce réglage de la probabilité de libération.
Les détails ne sont pas anodins. Un agoniste partiel comme Δ9-THC peut ne pas reproduire le profil complet produit par une brève poussée endogène de 2-AG. Un agoniste complet synthétique ne préservera pas nécessairement non plus la temporalité physiologique. La dose compte. Le réservoir de récepteurs aussi. Dans une synapse présentant une forte densité de CB1, même un agoniste partiel peut produire un effet important sur la libération de neurotransmetteur. Dans un autre tissu, le même ligand peut paraître faible.
CB2 a une physiologie synaptique directe moins bien établie que CB1, mais il réduit aussi l’AMPc et peut réguler la signalisation calcique, les voies des kinases et la libération de médiateurs inflammatoires dans les cellules immunitaires et gliales. Cela rend CB2 pertinent pour la communication neurone-glie, surtout dans les états pathologiques où l’expression du récepteur change. L’article d’analyse de réseau indexé dans PubMed en 2025/2026 a considéré CB1 et CB2 comme des nœuds influents dans des réseaux plus larges de signalisation endocannabinoïde et métabolique, ce qui constitue un meilleur cadre que de les traiter comme des interrupteurs isolés.
Désensibilisation, internalisation et régulation des récepteurs
Comment les récepteurs s’adaptent à une exposition répétée à des agonistes
- Phosphorylation Les régions intracellulaires du récepteur sont modifiées par les kinases des GPCR et d’autres kinases.
- Recrutement des bêta-arrestines Les arrestines découplent les récepteurs des protéines G et peuvent initier des signalisations supplémentaires.
- Désensibilisation Le récepteur devient moins réactif.
- Internalisation Les récepteurs sont internalisés dans des voies endocytaires.
- Sort après internalisation Les récepteurs peuvent être recyclés vers la membrane ou être dégradés.
Aucun récepteur ne peut être activé de façon continue sans conséquences. Pour CB1 et CB2, une exposition prolongée ou répétée à un agoniste conduit généralement à la phosphorylation des régions intracellulaires du récepteur par des kinases GPCR et d’autres kinases, au recrutement de β-arrestines, au découplage des protéines G, puis à l’internalisation via des voies endocytiques. La désensibilisation survient d’abord. L’endocytose suit souvent. Le recyclage ou la dégradation viennent ensuite.
Pour CB1, ce cycle régulateur est une raison majeure pour laquelle les effets aigus et chroniques diffèrent. Les agonistes puissants peuvent déclencher une désensibilisation rapide dans les systèmes cellulaires et une tolérance mesurable in vivo. La régulation spécifique aux régions cérébrales compte ici. Les récepteurs CB1 ne se désensibilisent pas de manière égale dans toutes les populations neuronales, ce qui aide à expliquer pourquoi la tolérance se développe de manière inégale selon les effets des cannabinoïdes. Les réponses analgésiques, l’hypothermie, les troubles de la mémoire et les effets moteurs peuvent évoluer à des rythmes différents parce que le récepteur est régulé différemment selon les circuits.
Les β-arrestines ne sont pas seulement des mécanismes d’arrêt. Elles peuvent servir d’échafaudage à leurs propres cascades de signalisation, notamment les voies MAP kinase, raison pour laquelle le recrutement des arrestines est devenu central dans l’agonisme biaisé. Un ligand qui inhibe fortement l’AMPc mais recrute faiblement la β-arrestine peut se comporter différemment d’un ligand qui fait les deux efficacement. Ce n’est plus un détail théorique ; c’est une stratégie de conception de médicaments. La discussion de 2025 dans le American Journal of Psychiatry sur le biais de CB1 dans la schizophrénie reflète une leçon plus large des GPCR : éviter certaines branches de signalisation peut réduire certaines limites, mais la sélectivité pour une voie ne garantit pas le succès clinique.
L’internalisation elle-même dépend aussi du ligand. Certains agonistes provoquent une endocytose étendue des récepteurs ; d’autres entraînent une internalisation limitée malgré l’activation des protéines G. Les modulateurs allostériques compliquent davantage le tableau en modifiant la manière dont les ligands orthostériques stabilisent les états du récepteur. C’est ici que la pharmacologie structurale rencontre la thérapeutique. La revue structurale de 2026 a clairement montré que la conformation du récepteur contrôle ensemble l’efficacité de la signalisation et la régulation du récepteur, et non comme des sujets séparés.
C’est la leçon centrale de signalisation à retenir. CB1 et CB2 ne sont pas de simples détecteurs marche-arrêt pour les cannabinoïdes. Ce sont des nœuds régulés dont la sortie change à l’échelle de la milliseconde au jour. Toute tentative sérieuse de les cibler, que ce soit pour l’épilepsie, la douleur, la psychose ou les maladies inflammatoires, doit prendre en compte le couplage Gi/o, les seconds messagers, le contrôle des canaux ioniques, la localisation synaptique et le fait que le récepteur s’adaptera au fait d’être stimulé.
Signalisation biaisée : pourquoi un seul récepteur peut produire différents résultats biologiques
L’ancienne vision de la pharmacologie du cannabinoid traitait un récepteur comme un interrupteur : les agonistes l’activent, les antagonistes le désactivent, et tout le reste dépend de l’endroit où ce récepteur est exprimé. Cette vision n’est pas suffisante pour CB1 ou CB2. Elle ne permet pas d’expliquer pourquoi deux ligands agissant sur le même récepteur peuvent produire des effets comportementaux, cognitifs, inflammatoires ou thérapeutiques très différents. Elle n’explique pas non plus pourquoi la découverte de médicaments autour des récepteurs cannabinoïdes a produit à plusieurs reprises des composés prometteurs in vitro mais finalement décevants, mal tolérés ou cliniquement ambigus.
Cela dépasse largement la théorie académique des récepteurs. L’Organisation mondiale de la Santé a estimé que 200 millions de personnes ont utilisé cannabis en 2019, soit environ 4 % de la population mondiale âgée de 15 à 64 ans. La schizophrénie touche environ 24 millions de personnes dans le monde. Dans ce contexte, la pharmacologie de CB1 n’est pas une question de niche. Elle se situe à l’intersection de la santé publique, de la psychiatrie et de la conception de médicaments. La FDA américaine, en 2025, avait approuvé un seul médicament dérivé du cannabis et trois médicaments liés au cannabis, soit un nombre très limité au regard de l’ampleur de l’intérêt clinique. L’une des raisons pour lesquelles les progrès ont été plus lents que ne le suggère le débat public est que la signalisation des récepteurs cannabinoïdes ne se réduit pas à une simple occupation du récepteur. Il s’agit d’une sélection de voies.
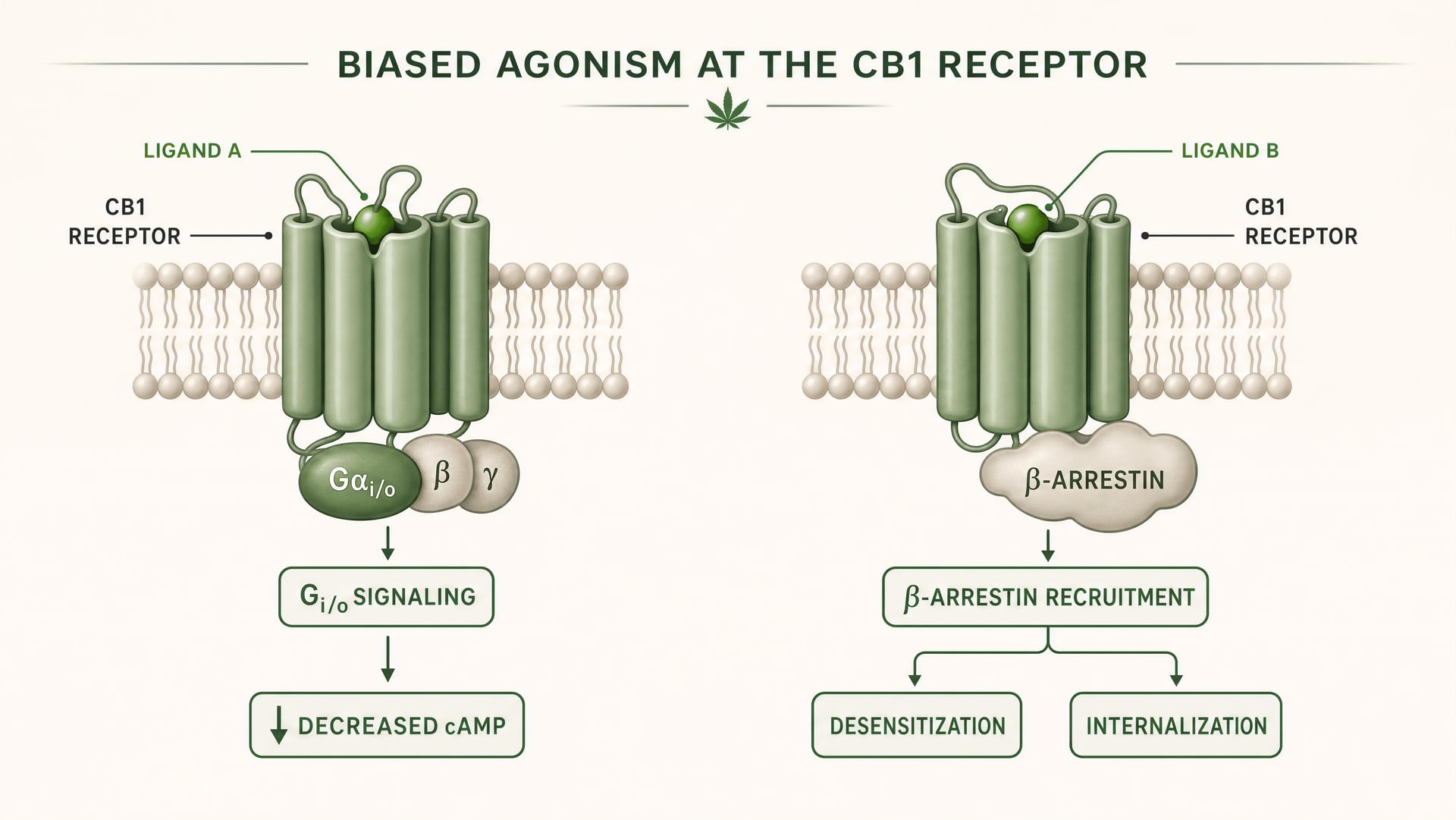
Ce que signifie l’agonisme biaisé en pharmacologie des GPCR
CB1 et CB2 sont des récepteurs couplés aux protéines G de classe A. Les travaux fondateurs d’Allyn Howlett ont établi CB1 comme un récepteur cannabinoid couplé à Gi/o, contribuant à faire passer le domaine d’une pharmacologie floue à des mécanismes définis par récepteur. Mais le couplage à Gi/o n’est que le début de l’histoire. Une fois qu’un ligand se lie, le récepteur peut adopter plus d’une forme active, et ces formes ne signalent pas de manière identique. Certaines conformations du récepteur favorisent l’activation des protéines G. D’autres recrutent plus fortement les beta-arrestins. Certains états favorisent la phosphorylation du récepteur, sa désensibilisation ou son internalisation. D’autres produisent une signalisation plus durable depuis la membrane plasmique ou depuis des compartiments endosomaux.
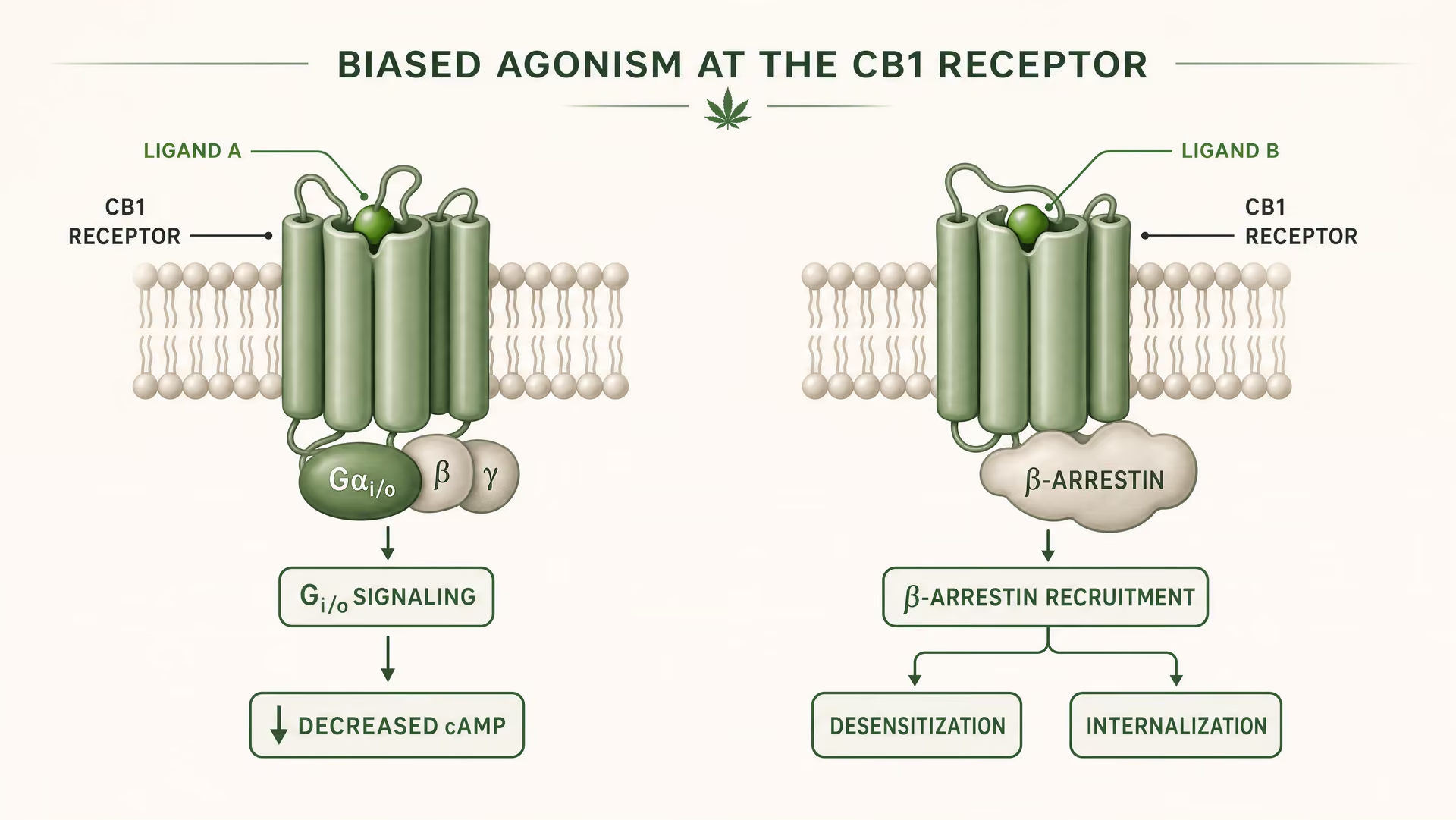{kind=link}
Agonisme biaisé Propriété d’un ligand qui favorise certaines voies de signalisation en aval plutôt que d’autres au même récepteur.
En termes simples, c’est cela, l’agonisme biaisé : différents ligands stabilisent différentes conformations du récepteur, et ces conformations favorisent différentes voies de signalisation en aval. Un récepteur n’est pas simplement actif ou inactif. Il est orienté conformationnellement.
Pour CB1, cela est particulièrement important parce que le récepteur se situe dans un environnement de signalisation dense, plastique et fortement dépendant du type cellulaire. Dans une terminaison glutamatergique corticale, un ligand peut réduire la libération du neurotransmetteur via l’inhibition de l’adénylate cyclase médiée par Gi/o et la modulation des canaux ioniques. Dans un interneurone GABAergique, le même récepteur peut déplacer l’équilibre local des circuits dans une direction très différente. Si le ligand favorise aussi un recrutement important des beta-arrestins, le récepteur peut internaliser plus rapidement, écourtant un effet tout en en ouvrant un autre. Le temps change. La localisation du signal change. Le résultat physiologique change.
Il ne s’agit pas d’une subtilité théorique. La revue structurelle de 2026 dans Frontiers in Chemical Biology sur les récepteurs cannabinoïdes le souligne clairement : la sélectivité des ligands pour CB1 et CB2 dépend de différences structurales au niveau du récepteur qui modifient la liaison, l’efficacité de la signalisation et la régulation du récepteur. Le mot clé ici est régulation. Un ligand peut présenter une affinité similaire tout en différant par son efficacité, son recrutement des arrestines, son temps de résidence ou sa propension à déclencher la désensibilisation. L’étude indexée dans PubMed de 2025/2026 sur le mécanisme dynamique de la sélectivité des sous-types pousse cette idée plus loin en soutenant que la sélectivité émerge de la dynamique conformationnelle, et non d’un modèle statique de type clé-serrure. Les endocannabinoids, les phytocannabinoids et les ligands synthétiques ne doivent donc pas être mis dans le même panier. L’anandamide, découverte par Raphael Mechoulam et Lumír Hanuš, ne se comporte pas comme delta-9-tetrahydrocannabinol, et aucun des deux ne se comporte comme une sonde synthétique hautement optimisée.
La signalisation biaisée explique aussi pourquoi les modulateurs allostériques suscitent autant d’intérêt. Un ligand allostérique peut ne pas activer directement CB1 comme le ferait un agoniste orthostérique, mais il peut remodeler les préférences de signalisation du récepteur, en amplifiant une voie et en en atténuant une autre. Cela ouvre une voie vers un contrôle fin. En théorie.
La signalisation biaisée de CB1 comme piste de recherche sur la schizophrénie
L’article de 2025 dans l’American Journal of Psychiatry avance l’un des arguments récents les plus solides selon lequel la signalisation biaisée de CB1 n’est pas seulement un concept de pharmacologie, mais une stratégie thérapeutique plausible pour la schizophrénie. Cet argument mérite attention, car la recherche sur la schizophrénie a généralement abordé les cannabinoids sous l’angle de l’épidémiologie, de l’association avec le risque ou d’avertissements généraux concernant la psychose. L’article de l’AJP déplace le cadre. Il demande si le problème n’est pas « les cannabinoids » en général, ni même « l’activation de CB1 » en général, mais quelles formes de signalisation de CB1 sont engagées, dans quels circuits, et pendant combien de temps.
C’est une bien meilleure question.
CB1 est l’un des GPCR les plus abondants dans le cerveau, avec une forte expression dans le cortex, l’hippocampe, les ganglions de la base et le cervelet, mais cette abondance n’explique pas à elle seule les effets cliniques. La schizophrénie implique une dérégulation de la saillance, de la cognition, de la perception et de la coordination des réseaux à travers les systèmes corticaux et sous-corticaux. Un récepteur capable de moduler l’activité des circuits glutamatergiques, GABAergiques et liés à la dopamine est donc pertinent par conception. L’article de l’AJP soutient que des ligands CB1 biaisés pourraient dissocier des effets thérapeutiques utiles sur les circuits de certaines toxicités, telles que l’altération cognitive, l’anxiété, la dysphorie ou des réponses psychotomimétiques.
C’est une affirmation ambitieuse, mais elle n’est pas spéculative. Elle s’inscrit dans le prolongement du domaine plus large des GPCR, où le biais des voies a déjà changé la manière dont les chercheurs envisagent les médicaments ciblant les récepteurs opioïdes, de l’angiotensine et de la dopamine. L’espoir translationnel autour de CB1 est que certaines sorties de signalisation puissent améliorer la fonction des réseaux corticaux ou atténuer des états de circuit aberrants sans reproduire le profil indésirable complet associé à un agonisme CB1 à forte efficacité.
La schizophrénie constitue un bon cas d’épreuve parce que l’exigence clinique est élevée. Un candidat médicament ne peut pas simplement modifier le comportement dans un modèle murin. Il doit éviter d’aggraver la psychose, la sédation et les troubles cognitifs chez des personnes déjà vulnérables à ces problèmes. Cela fait immédiatement de la sélectivité de voie bien plus qu’une préférence de chimie médicinale. Elle devient une exigence de sécurité.
Le cadre proposé par l’AJP permet aussi de corriger une simplification fréquente dans les discussions sur cannabis. Delta-9-THC est un phytocannabinoid avec une activité agoniste partielle sur CB1, mais ses effets reflètent la dose, le timing, la réserve de récepteurs, le tonus local de l’endocannabinoid et l’engagement des voies dans différentes populations neuronales. Un ligand synthétique de CB1 conçu pour favoriser une voie intracellulaire particulière pourrait se comporter de manière très différente de THC, même si tous deux « ciblent CB1 ». L’inverse est également vrai : deux composés qui améliorent tous deux un critère préclinique pertinent pour la schizophrénie peuvent diverger fortement sur la cognition ou l’affect si l’un déclenche une signalisation dominée par les arrestines et l’autre non. L’identité du récepteur ne permet pas à elle seule de prédire l’ensemble du phénotype.
Pourquoi la sélectivité de voie compte pour la sécurité et l’efficacité
La sélectivité de voie compte parce que l’efficacité n’est pas une dimension unique. Un médicament cannabinoid peut être puissant et néanmoins cliniquement médiocre. Il peut être sélectif pour CB1 et échouer malgré tout. Il peut éviter CB2 et produire malgré tout des effets immunologiques ou métaboliques indésirables par interactions en réseau. L’analyse de réseau intégrative indexée dans PubMed en 2025/2026 a identifié CB1 et CB2 comme des nœuds très influents du système endocannabinoid et a cartographié leur signalisation sur des voies métaboliques. Cette vision systémique est essentielle. Les récepteurs ne fonctionnent pas isolément, et le biais de voie à un nœud peut se répercuter sur des programmes physiologiques plus larges.
Pour CB1, les préoccupations de sécurité sont évidentes. Une forte activation centrale de CB1 peut provoquer des troubles de la mémoire, une altération de la perception, de l’anxiété, une tachycardie et, chez les personnes susceptibles, des effets liés à la psychose. Tout programme thérapeutique visant la douleur, l’appétit, l’humeur, l’addiction ou la schizophrénie doit affronter ce profil de risques. Un ligand qui conserve un effet synaptique désiré médié par Gi/o tout en limitant la désensibilisation ou d’autres cascades de signalisation indésirables liées aux beta-arrestins pourrait, en théorie, élargir la fenêtre thérapeutique. Mais « en théorie » est important. De nombreux programmes de ligands biaisés en pharmacologie des GPCR ont montré qu’un biais mesuré dans un système d’essai donné ne prédit pas toujours les résultats in vivo. Le contexte cellulaire, la densité des récepteurs, l’expression des effecteurs et la cinétique peuvent tous modifier le biais apparent.
CB2 offre un parallèle de prudence. La revue de 2026 dans Frontiers in Behavioral Neuroscience décrit une mise à jour sur les 3 dernières années, au cours de laquelle la signalisation de CB2 a attiré l’attention dans les troubles du système nerveux central en raison de liens avec des mécanismes neuroinflammatoires et neurodégénératifs. Cela contredit directement l’ancienne idée selon laquelle CB2 serait sans pertinence pour le cerveau. Cela dit, cibler simplement CB2 ne garantit pas un médicament anti-inflammatoire utile. La distribution est plus graduée que l’ancienne séparation cerveau-versus-corps, et les conséquences de la signalisation dépendent toujours du ligand et du contexte.
La leçon pratique est donc claire : la sélectivité pour le sous-type de récepteur est nécessaire, mais pas suffisante. La sélectivité de voie peut faire la différence entre un cannabinoid qui semble thérapeutique, un autre qui est intoxicant, et un troisième qui échoue en essai parce qu’il ne parvient pas à séparer bénéfice et effet indésirable. Pour CB1, surtout en psychiatrie, cette distinction décidera probablement si le récepteur reste un avertissement ou devient une cible médicamenteuse viable.
Biologie structurale de CB1 et CB2 : comment la forme détermine la sélectivité
La biologie structurale a changé la manière dont les récepteurs cannabinoïde sont abordés. L’ancienne formule simplificatrice — CB1 explique l’intoxication, CB2 explique l’inflammation — ignore le fait que les deux récepteurs sont des récepteurs couplés aux protéines G de classe A dont le comportement dépend de la forme, du mouvement, de la profondeur de liaison et des partenaires de signalisation disponibles dans une cellule donnée. Cela a une portée bien au-delà de la pharmacologie de base. On estime que le cannabis a été utilisé par 200 millions de personnes en 2019, soit 4 % de la population mondiale âgée de 15 à 64 ans, selon l’WHO, pourtant la FDA ne répertorie encore en 2025 qu’un seul produit médicamenteux dérivé du cannabis et trois produits médicamenteux liés au cannabis comme approuvés. L’une des raisons de cet écart est structurelle : il est difficile de concevoir des ligands cannabinoïde qui ciblent le bon récepteur, de la bonne manière, pendant la bonne durée.
La revue de Frontiers in Chemical Biology de 2026 le montre clairement. CB1 et CB2 ne diffèrent pas seulement par leur lieu d’expression. Ils diffèrent par l’architecture de leurs poches de liaison des ligands, par la forme et la flexibilité de leurs boucles extracellulaires, par l’agencement de leurs hélices transmembranaires et par les états conformationnels qu’ils privilégient après la liaison d’un ligand. Ces caractéristiques influencent non seulement la sélectivité, mais aussi l’efficacité, la désensibilisation, l’internalisation et la sélectivité de voie.
Ce que les études structurales révèlent à propos des poches des récepteurs
Une poche orthostérique est la principale cavité de liaison où des ligands endogènes tels que l’anandamide et le 2-arachidonoylglycérol, des phytocannabinoïdes tels que THC, ainsi que de nombreux ligands synthétiques établissent leur contact principal. Dans CB1 et CB2, cette poche se situe au sein du faisceau des sept hélices transmembranaires, partiellement coiffé par des boucles extracellulaires qui peuvent soit en ouvrir l’accès, soit le restreindre.
Les structures obtenues par cryo-EM et rayons X au cours des dernières années ont montré que les récepteurs cannabinoïde ne fonctionnent pas comme des serrures rigides attendant une clé. Il est plus juste de les comprendre comme des cibles mobiles dotées de formes préférentielles. La revue de 2026 dans Frontiers in Chemical Biology souligne que les cavités orthostériques de CB1 et CB2 sont suffisamment similaires pour lier des classes de ligands qui se recoupent, mais suffisamment différentes en taille, en identité de résidus et en flexibilité locale pour modifier l’affinité et l’issue de signalisation. C’est pourquoi des composés étroitement apparentés peuvent se distinguer pharmacologiquement. Une petite variation de l’encombrement d’un substituant, de la polarité ou de la longueur de la chaîne peut modifier la profondeur de pénétration d’un ligand dans la poche, les hélices qu’il pousse et la manière dont le récepteur se stabilise dans un état favorisant la protéine G ou favorisant l’arrestine.
CB1 a été particulièrement instructif sur le plan structural, car de nombreux modèles à haute résolution à l’état inactif et à l’état actif existent désormais. Un thème récurrent est que sa poche est vaste et hydrophobe, ce qui correspond à la nature lipophile de nombreux cannabinoïde. La boucle extracellulaire 2 et les parties supérieures de plusieurs hélices contribuent à façonner la zone d’entrée. Les hélices transmembranaires sont les sept segments traversant la membrane qui constituent le cœur du récepteur ; lorsqu’un ligand se lie, ces hélices peuvent se déplacer les unes par rapport aux autres. Le mouvement le plus important sur le plan pharmacologique se situe généralement du côté intracellulaire, où le déplacement vers l’extérieur de l’hélice 6 contribue à créer un site d’ancrage pour les protéines Gi/o. Ce déplacement est l’un des signes caractéristiques de l’activation du récepteur.
CB2 partage le même repliement général des GPCR, mais la revue Frontiers soutient que les différences spécifiques à la sous-type dans les acides aminés autour de la poche et des régions de boucle offrent aux chimistes médicinaux des leviers exploitables pour la sélectivité. Il ne s’agit pas de dire qu’une poche serait simplement « cérébrale » et l’autre « immunitaire ». La question est géométrique et énergétique. Des résidus distincts modifient le contour de la cavité, les possibilités locales de liaison hydrogène, l’empilement aromatique et la flexibilité des canaux d’accès par lesquels les ligands entrent depuis la membrane.
Une étude indexée dans PubMed en 2025/2026 sur le mécanisme dynamique de la sélectivité entre sous-types a poussé l’analyse plus loin en avançant que la sélectivité des endocannabinoid ne dépend pas seulement d’une affinité de liaison statique. Les dynamiques conformationnelles comptent. En termes simples, un récepteur peut explorer plusieurs formes avant et après la liaison du ligand, et certains ligands stabilisent mieux qu’autres une forme sélective. Cela aide à expliquer pourquoi les lipides endogènes, les phytocannabinoïdes et les ligands synthétiques peuvent montrer des préférences de sous-type différentes, même lorsque leurs charpentes paraissent proches sur le papier.
Déterminants de la sélectivité des ligands entre CB1 et CB2
La sélectivité commence par la chimie des contacts, mais ne s’y limite pas. La revue de Frontiers in Chemical Biology présente la sélectivité comme le produit de différences structurales au niveau du récepteur qui affectent à la fois la liaison, l’efficacité de signalisation et la régulation. C’est le bon cadre d’analyse. Un ligand peut être sélectif de CB2 dans un test de liaison par radioligand et perdre son avantage pratique s’il induit aussi des états du récepteur qui provoquent une tolérance rapide ou une signalisation faible dans des cellules pertinentes pour la maladie.
Plusieurs caractéristiques structurales reviennent de façon répétée. Premièrement, la composition en acides aminés de la poche orthostérique diffère suffisamment entre CB1 et CB2 pour modifier la manière dont le groupement tête, le noyau et la chaîne hydrophobe d’un ligand sont accommodés. Deuxièmement, les boucles extracellulaires contribuent à la forme de l’entrée et à l’orientation. Troisièmement, les régions supérieures et intermédiaires des hélices transmembranaires peuvent orienter le récepteur vers des ensembles d’états actifs légèrement différents. Un état conformationnel n’est rien d’autre qu’une des formes possibles du récepteur à un moment donné. Les différents ligands ne se contentent pas de se lier à un récepteur ; ils stabilisent un sous-ensemble de ces formes.
C’est pourquoi la sélectivité de sous-type est souvent modeste pour les cannabinoïdes naturels. THC, par exemple, agit sur les deux récepteurs. L’anandamide et le 2-AG agissent aussi sur les deux, bien qu’avec des différences dépendantes du contexte en puissance, efficacité et métabolisme. Les ligands synthétiques ont été plus utiles pour disséquer les relations structure-sélectivité, car les chimistes peuvent modifier systématiquement des caractéristiques telles que la longueur de chaîne latérale, les contraintes cycliques et les substituants polaires. Même dans ce cas, une séparation nette reste difficile. CB1 et CB2 sont suffisamment homologues pour qu’un composé conçu pour l’un conserve souvent une activité significative sur l’autre.
Cela a des conséquences pratiques. Les développeurs de médicaments poursuivent depuis longtemps des agonistes sélectifs de CB2 dans l’espoir d’obtenir des effets anti-inflammatoires ou analgésiques sans effets indésirables centraux marqués médiés par CB1. Parfois, cette stratégie fonctionne pharmacologiquement, mais ce n’est pas un laissez-passer. La revue de 2026 dans Frontiers in Behavioral Neuroscience souligne que CB2 a suscité de l’attention dans les troubles du système nerveux central au cours des « 3 dernières années », ce qui contredit l’idée simple selon laquelle CB2 serait sans pertinence pour le cerveau. Ainsi, même un ligand CB2 « périphérique » ne peut pas être interprété à l’aide de cartes récepteur dépassées.
Pourquoi l’efficacité et la régulation sont aussi des questions structurales
La puissance répond à la question de la quantité de ligand nécessaire. L’efficacité demande ce que ce ligand fait au récepteur une fois lié. La biologie structurale relie les deux, sans les confondre. Deux ligands peuvent occuper la même poche et produire des réponses très différentes parce qu’ils stabilisent des conformations actives différentes.
Pour CB1, cela est au cœur de la réflexion thérapeutique actuelle. L’article de American Journal of Psychiatry de 2025 soutient que la signalisation biaisée sur CB1 pourrait constituer une stratégie plausible pour la schizophrénie, un trouble qui touche environ 24 millions de personnes dans le monde. L’agonisme biaisé signifie qu’un ligand favorise une voie en aval plutôt qu’une autre, souvent la signalisation Gi/o plutôt que le recrutement de beta-arrestin, ou l’inverse. Sur le plan structurel, ce biais provient du fait que le ligand déplace la face intracellulaire du récepteur vers une forme qui s’ajuste mieux à un partenaire de signalisation qu’à un autre. Ce n’est pas une idée abstraite. C’est une cible de chimie médicinale.
Les beta-arrestins sont importants parce qu’ils contribuent à la désensibilisation et à l’internalisation. La désensibilisation signifie que le récepteur devient moins réactif après une activation répétée ou prolongée. L’internalisation signifie que le récepteur est retiré de la membrane de surface vers l’intérieur de la cellule. Ces deux processus sont influencés par la conformation du récepteur. Certains ligands activent fortement les protéines G mais recrutent faiblement les arrestines ; d’autres font les deux. Un médicament CB1 tolérable sur le plan comportemental pourrait nécessiter exactement ce type de dissociation, plutôt qu’un simple blocage ou un agonisme complet.
La modulation allostérique ajoute un niveau supplémentaire. Un ligand allostérique se lie en dehors de la poche orthostérique et modifie la réponse du récepteur à d’autres ligands. Sur le plan structural, cette approche est séduisante parce qu’elle peut permettre un contrôle plus fin de la forme du récepteur qu’une stimulation brutale de la poche principale avec un agoniste à forte efficacité. En pharmacologie des récepteurs cannabinoïde, cela pourrait signifier préserver une signalisation utile tout en réduisant les effets indésirables liés à une activation excessive de CB1.
La leçon générale est sans détour. La forme du récepteur n’est pas un détail secondaire. C’est l’explication du fait que les phytocannabinoïdes, les endocannabinoid et les composés synthétiques peuvent produire des profils cliniques différents même lorsqu’ils ciblent la même famille de récepteurs. Comme CB1 et CB2 occupent des nœuds influents dans les analyses de réseaux de l’endocannabinoid-system, comme l’a rapporté une étude systémique indexée dans PubMed en 2025/2026, des erreurs de sélectivité ou d’efficacité peuvent se propager à de nombreuses voies. L’information structurale ne garantit pas le succès d’un médicament. Elle explique en revanche pourquoi il reste si difficile d’aligner sélectivité, puissance et tolérance.
Endocannabinoïdes et sélectivité des sous-types : anandamide, 2-AG, et comportement dynamique des récepteurs
La pharmacologie des endocannabinoïdes est souvent expliquée à l’aide d’un raccourci qui paraît clair, mais qui induit en erreur : l’anandamide et le 2-arachidonoylglycérol (2-AG) sont traités comme s’il s’agissait des « agonistes standards » intégrés de l’organisme, utiles surtout comme points de référence pour ce que font normalement CB1 et CB2. Ce cadrage manque l’essentiel. Il ne s’agit pas d’hormones stables circulant librement et rencontrant les récepteurs dans des conditions uniformes. Ce sont des médiateurs lipidiques à courte durée de vie, produits à la demande, dans les membranes, par des systèmes enzymatiques spécifiques à chaque cellule, puis rapidement détruits. Leur préférence apparente pour certains récepteurs peut varier selon la composition membranaire, les conformations des récepteurs, l’activité enzymatique locale et le moment de leur libération.
Cela a de l’importance au-delà de la théorie académique des récepteurs. Selon la WHO, en 2019, le cannabis était utilisé par environ 200 millions de personnes, soit 4 % de la population mondiale âgée de 15 à 64 ans, et les médicaments ciblant les cannabinoid sont déjà utilisés en clinique : la FDA a indiqué en 2025 avoir approuvé un médicament dérivé du cannabis et trois médicaments liés au cannabis. Si les ligands endogènes ne se comportent pas comme de simples témoins de manuels, alors la comparaison des phytocannabinoid ou des candidats médicaments avec eux n’a rien d’évident.
Pourquoi les ligands endogènes ne sont pas de simples composés de référence
L’anandamide, identifiée en 1992 par Devane, Hanus, Breuer, Mechoulam et leurs collègues, et le 2-AG, établi quelques années plus tard comme un autre endocannabinoïde majeur, diffèrent bien au-delà des tableaux de puissance. Ils proviennent de voies biosynthétiques différentes, atteignent les récepteurs selon des échelles de temps différentes et sont éliminés par des enzymes différentes. L’anandamide est générée principalement à partir de la N-arachidonoyl phosphatidyléthanolamine par des voies impliquant NAPE-PLD, bien que des voies alternatives existent. Le 2-AG est produit en grande partie à partir du diacylglycérol par DAGLα et DAGLβ. Ce ne sont pas des notes biochimiques mineures ; elles déterminent où et quand l’activation des récepteurs se produit.
Même l’affirmation plus ancienne selon laquelle l’anandamide serait un ligand préférentiellement dirigé vers CB1 tandis que le 2-AG serait un agoniste complet de CB1 et CB2 peut devenir trop simpliste lorsqu’elle est transposée de systèmes purifiés à des tissus vivants. L’anandamide est également un substrat de la fatty acid amide hydrolase, ou FAAH, et peut activer des cibles non cannabinoid telles que TRPV1 dans certaines conditions. Le 2-AG est présent dans le tissu cérébral à des niveaux de base beaucoup plus élevés que l’anandamide dans de nombreuses mesures et est hydrolysé principalement par la monoacylglycérol lipase, ou MAGL, avec une contribution également de ABHD6 et ABHD12. Ainsi, l’« entrée » que chaque ligand fournit au récepteur ne dépend pas seulement de l’affinité ou de l’efficacité sur CB1 versus CB2. Elle dépend du fait que le ligand soit produit à la synapse, dans une cellule immunitaire, près d’une membrane intracellulaire, ou dans un compartiment riche en enzymes de dégradation.
Les travaux d’Allyn Howlett sur la pharmacologie des récepteurs ont contribué à établir CB1 comme un véritable GPCR couplé principalement aux protéines Gi/o. Ce cadre reste essentiel, mais il ne permet plus de soutenir un modèle simple de ligand endogène comme médicament de référence. CB1 et CB2 peuvent signaler via l’inhibition de l’adénylate cyclase, la régulation des canaux ioniques, les voies MAP kinase, le recrutement de beta-arrestin, l’internalisation des récepteurs et la désensibilisation. Un ligand qui paraît « plus faible » dans un essai peut produire une signalisation plus prolongée dans un tissu où la dégradation est lente, ou favoriser une voie de signalisation en aval plutôt qu’une autre. L’article de 2025 du American Journal of Psychiatry sur la signalisation biaisée de CB1 souligne ce point dans un contexte pathologique : la question thérapeutique dans des troubles tels que la schizophrénie, qui touche environ 24 millions de personnes dans le monde, n’est pas seulement de savoir si CB1 est activé, mais comment.
Mécanismes dynamiques de sélectivité des sous-types
L’étude récente sur la sélectivité des sous-types, indexée dans PubMed (PMID: 41962866), est importante parce qu’elle éloigne le domaine d’une logique de type serrure-et-clé. Son message central est que la sélectivité des endocannabinoïdes peut résulter de mécanismes dynamiques. Autrement dit, la préférence pour un sous-type de récepteur ne s’explique pas seulement par une différence fixe dans la force de liaison d’un ligand à CB1 par rapport à CB2. Le complexe ligand-récepteur explore plusieurs états conformationnels, et ces états diffèrent entre CB1 et CB2.
Cette observation concorde avec la revue de 2026 dans Frontiers in Chemical Biology, qui soutient que les différences structurales entre CB1 et CB2 façonnent la liaison, l’efficacité de signalisation et la régulation des récepteurs. Les récepteurs présentent une similarité de séquence importante, mais de petites différences dans l’architecture de la poche orthostérique, les boucles extracellulaires, les mouvements des hélices transmembranaires et les surfaces de couplage intracellulaire peuvent orienter ce qui se produit après la liaison du ligand. Un ligand peut pénétrer dans des poches similaires sur les deux récepteurs mais stabiliser un état plus compétent pour la signalisation dans un sous-type, ou favoriser des transitions différentes entre les états inactif, intermédiaire et actif.
Pour les endocannabinoïdes, cela est particulièrement plausible parce qu’il s’agit de lipides flexibles plutôt que d’échafaudages synthétiques rigides. La flexibilité est une variable pharmacologique. L’anandamide peut adopter plusieurs conformations, et le 2-AG est tout aussi dynamique dans les membranes. La question devient alors non seulement « Se lie-t-il ? », mais « Quels états du récepteur favorise-t-il, combien de temps ces états sont-ils occupés, et quelles protéines sont disponibles pour le couplage suivant ? » Un neurone riche en CB1 et une cellule microgliale exprimant CB2 ne présentent pas le même environnement de signalisation, même avant de prendre en compte les différences de densité des récepteurs.
Cette vision dynamique affaiblit également l’ancienne hypothèse selon laquelle les ligands endogènes définissent une seule base physiologique à partir de laquelle THC, cannabidiol ou les cannabinoid de synthèse peuvent être classés. THC est un phytocannabinoid avec son propre profil d’efficacité et son propre comportement cinétique. Les endocannabinoïdes sont des signaux dépendants de l’événement, souvent phasiques et locaux. Un agoniste synthétique de CB2 peut sembler sélectif dans un test recombinant, mais produire des résultats inattendus dans un tissu si la réserve de récepteurs, le cholestérol membranaire, la formation d’hétéromères ou la gestion par beta-arrestin diffèrent. La sélectivité sur le papier n’est pas un destin.
Comment la synthèse et la dégradation locales façonnent l’engagement des récepteurs
In vivo, la signalisation des endocannabinoïdes est gouvernée autant par l’enzymologie que par la pharmacologie des récepteurs. Dans de nombreuses synapses centrales, le 2-AG est produit au niveau postsynaptique en réponse à des élévations de calcium ou à l’activation de récepteurs couplés à Gq/11, puis se déplace rétrogradement vers les récepteurs CB1 présynaptiques et supprime la libération de neurotransmetteurs. Il s’agit d’un mécanisme temporel mesuré en secondes, et non d’un tonus de fond diffus. Le signal prend fin lorsque MAGL et des hydrolases apparentées éliminent le 2-AG. L’anandamide se comporte souvent différemment, avec une abondance tissulaire plus faible, des conditions de libération distinctes et une sensibilité accrue à la dégradation contrôlée par FAAH.
Cela signifie que la modification d’une enzyme peut altérer l’engagement des récepteurs sans modifier les récepteurs eux-mêmes. L’inhibition de FAAH augmente le tonus de l’anandamide, mais le résultat physiologique dépend du lieu de production de l’anandamide, de la présence éventuelle de TRPV1, et du fait que les récepteurs CB1 dans ce microdomaine soient désensibilisés ou internalisés. L’inhibition de MAGL peut élever fortement le 2-AG, mais une élévation chronique peut conduire à une désensibilisation de CB1 dans certains systèmes. Davantage de ligand ne signifie pas toujours une signalisation plus utile.
L’étude de biologie des systèmes indexée sous PMID: 42129940 renforce cette vision plus large en plaçant CB1 et CB2 parmi des nœuds influents reliés à des voies métaboliques, et non comme des interrupteurs isolés. C’est l’échelle appropriée pour comprendre l’action des endocannabinoïdes. La disponibilité du ligand, l’accès membranaire, la conformation du récepteur, le couplage aux protéines G, le recrutement de beta-arrestin et la dégradation interagissent tous. Il en va de même pour les gradients tissulaires. CB2 n’est pas simplement « en dehors du cerveau », et CB1 n’est pas uniquement « le récepteur de la psychoactivité ». La signalisation des endocannabinoïdes est locale, conditionnelle et cinétique. Toute description de la sélectivité des sous-types qui ignore ces caractéristiques interprétera mal à la fois la physiologie et le développement de médicaments.
Le système endocannabinoïde comme réseau, et non comme un schéma à deux récepteurs
Le système endocannabinoïde n’a guère de sens s’il est représenté comme deux cercles étiquetés CB1 et CB2 avec des flèches pointant vers l’extérieur. Ce dessin simplifié a aidé le domaine à ses débuts, en particulier après les travaux de pharmacologie des récepteurs menés par Allyn Howlett et l’identification de l’anandamide par Raphael Mechoulam et Lumír Hanuš, mais il masque désormais davantage qu’il ne révèle. Les enjeux pratiques sont considérables. L’Organisation mondiale de la Santé a estimé que 200 millions de personnes avaient utilisé cannabis en 2019, soit environ 4 % de la population mondiale âgée de 15 à 64 ans, tandis que la FDA américaine indique qu’un médicament dérivé du cannabis et trois médicaments liés au cannabis sont approuvés en 2025. Un système de récepteurs qui touche autant d’expositions et suscite autant de revendications thérapeutiques ne peut pas être traité comme un simple duo « récepteur cérébral + récepteur immunitaire ».
Une approche systémique part d’un fait fondamental : les endocannabinoid sont des lipides produits à la demande, rapidement dégradés, et intégrés dans le même espace biochimique que les médiateurs de l’inflammation, les voies de remodelage membranaire et les boucles de rétroaction synaptique. L’anandamide et le 2-arachidonoylglycérol ne sont pas des « messages » isolés. Ils sont produits à partir de précurseurs membranaires, entrent en compétition avec d’autres voies lipidiques pour les substrats, et agissent dans des tissus où la densité des récepteurs, le type cellulaire, l’expression enzymatique et les partenaires de signalisation diffèrent tous. C’est pourquoi un même ligand peut paraître anxiolytique dans un contexte, intoxicant dans un autre, anti-inflammatoire ailleurs, et cliniquement décevant dans un essai.
Ce que l’analyse intégrative de réseau apporte
L’étude d’analyse intégrative de réseau indexée dans PubMed, publiée en 2025/2026 (PMID : 42129940), est utile parce qu’elle déplace l’attention des cibles isolées vers l’organisation du système. Au lieu de se demander seulement quel ligand se lie à CB1 ou CB2, les auteurs modélisent le système endocannabinoïde comme un réseau d’interactions incluant récepteurs, enzymes de biosynthèse et de dégradation, intermédiaires lipidiques et liens au niveau des voies vers l’inflammation, le métabolisme et la signalisation neuronale. Dans ce cadre, l’influence ne se définit pas seulement par l’abondance des récepteurs. Elle se définit par la connectivité et par la manière dont les perturbations se propagent dans le réseau.
Cela est déterminant pour l’interprétation. Si un médicament modifie la signalisation de CB1, l’effet en aval ne se limite pas à l’inhibition de l’adénylate cyclase médiée par Gi/o. Il peut modifier la libération des neurotransmetteurs, changer le comportement des canaux calciques et potassiques, modifier le recrutement de la beta-arrestine, déclencher l’internalisation des récepteurs et, indirectement, modifier le flux de substrats lipidiques qui alimentent aussi les eicosanoïdes et d’autres voies inflammatoires. Un modèle de réseau rend mieux compte de ces conséquences inter-systèmes qu’un tableau d’occupation des récepteurs.
C’est aussi là que l’ancienne simplification « CB1 dans le cerveau, CB2 dans les cellules immunitaires » montre ses limites. La distribution est graduée, spécifique aux cellules et dépendante de l’état physiologique. CB1 est abondant dans de nombreuses populations neuronales, mais pas uniformément dans le cerveau, et il apparaît aussi dans des tissus périphériques pertinents pour l’équilibre énergétique, la nociception et la fonction des organes. CB2 est enrichi dans les lignées immunitaires, mais des travaux plus récents sur le système nerveux central ont largement dépassé l’idée qu’il serait sans pertinence pour la biologie cérébrale. Une revue de 2026 dans Frontiers in Behavioral Neuroscience a décrit cela comme « une mise à jour des 3 dernières années », en mettant l’accent sur la signalisation de CB2 dans des contextes neuroinflammatoires et neurodégénératifs. Le point n’est pas que CB2 soit soudain devenu « aussi le récepteur cérébral ». Le point est que la signification d’un récepteur dépend des cellules qui sont actives, lésées, enflammées ou en adaptation.
L’analyse de réseau aide à expliquer pourquoi les médicaments ciblant les récepteurs produisent souvent des effets plus larges que prévu. Un phytocannabinoid actif sur CB1 comme le delta-9-THC, un endocannabinoid comme l’anandamide et un agoniste synthétique peuvent tous engager le même récepteur tout en produisant des résultats temporels et spatiaux différents. La classe du ligand compte. L’efficacité, le temps de résidence, le métabolisme et l’accès à différentes conformations du récepteur comptent aussi.
CB1 et CB2 comme nœuds influents
L’étude intégrative de réseau identifie CB1 et CB2 comme des nœuds hautement influents, non pas parce qu’ils agissent seuls, mais parce qu’ils occupent des points de contrôle où convergent de nombreuses voies. En termes de réseau, les nœuds influents sont des endroits où des changements locaux peuvent se propager largement. En pharmacologie, ce sont des récepteurs dont l’état d’activation peut réorienter en même temps la signalisation synaptique, les réponses immunitaires et les programmes cellulaires de stress.
CB1 est l’exemple le plus clair de la raison pour laquelle « influent » ne signifie pas « simple ». Il signale principalement via les protéines Gi/o, mais ce n’est qu’un début. Selon le ligand et le contexte, CB1 peut recruter la beta-arrestine, subir une désensibilisation dépendante de la phosphorylation, s’internaliser, se recycler ou rester activement présent à la membrane. Un article de l’American Journal of Psychiatry paru en 2025 soutient que la signalisation biaisée de CB1 constitue une stratégie thérapeutique plausible pour la schizophrénie, une maladie dont l’OMS indique qu’elle touche environ 24 millions de personnes dans le monde. Cette proposition n’a de sens que si CB1 est compris comme un hub de signalisation avec des sorties séparables, et non comme un simple interrupteur de l’intoxication. Un ligand qui favorise une branche de signalisation plutôt qu’une autre peut préserver certains effets thérapeutiques tout en réduisant les effets indésirables. Peut, et non pas va nécessairement. Le biais peut réduire les inconvénients, mais il n’efface pas le fait que CB1 s’inscrit dans des circuits qui gouvernent la cognition, la récompense, le contrôle moteur, le traitement de la douleur et l’appétit.
CB2 présente un problème similaire, mais en sens inverse. Il a longtemps été présenté comme la cible plus sûre en raison d’une association plus faible avec les effets psychoactifs aigus. Ce cadrage est trop optimiste. Les ligands dirigés vers CB2 peuvent néanmoins produire des résultats décevants ou mixtes si la signalisation pertinente à la maladie dépend de l’état cellulaire, de l’inductibilité du récepteur, du tonus inflammatoire ou des interactions croisées avec les enzymes métaboliques. La récente revue structurale de Frontiers in Chemical Biology, publiée en 2026, souligne que la sélectivité entre « CB1 et CB2 » dépend de différences structurales au niveau du récepteur qui modifient la liaison, l’efficacité et la régulation du récepteur. L’étude PubMed-indexée de 2025/2026 sur la sélectivité des sous-types (PMID : 41962866) va plus loin en avançant que la sélectivité est dynamique, façonnée par le comportement conformationnel plutôt que par un ajustement statique de type clé-serrure. C’est une bonne raison de cesser de parler de composés sélectifs pour CB1 ou CB2 comme si la sélectivité était une propriété fixe avec des conséquences biologiques fixes.
Liens entre signalisation des récepteurs et voies métaboliques
La contribution la plus précieuse de la perspective en réseau est qu’elle réunit à nouveau la pharmacologie des récepteurs et le métabolisme lipidique. La signalisation des endocannabinoid repose sur une chimie liée à l’acide arachidonique. L’anandamide et le 2-AG sont générés et dégradés par des systèmes enzymatiques qui influencent aussi la disponibilité des lipides bioactifs impliqués dans l’inflammation et la réponse tissulaire. Un médicament qui inhibe FAAH ou MAGL ne se contente pas de « faire augmenter les endocannabinoid ». Il redistribue le trafic de signalisation à travers un réseau métabolique.
Cela peut être utile. Cela peut aussi se retourner contre l’objectif recherché. L’augmentation du 2-AG, par exemple, peut renforcer la signalisation de CB1 ou de CB2 dans certains compartiments tout en modifiant des voies liées aux prostaglandines dans d’autres, par détournement des substrats ou oxydation en aval. L’implication clinique est simple : les stratégies ciblant les récepteurs et celles ciblant les enzymes peuvent avoir des conséquences à l’échelle du système même lorsque leur objectif de conception est étroit. C’est l’une des raisons pour lesquelles les composés sélectifs ne se sont pas automatiquement traduits par des profils cliniques nets dans la douleur, les maladies psychiatriques ou les troubles inflammatoires, malgré des besoins non satisfaits élevés. La douleur chronique touche à elle seule près d’un adulte sur cinq aux États-Unis, selon les National Academies, mais la pharmacologie du cannabis n’a pas produit de solution universellement fiable fondée sur les récepteurs.
Vu sous cet angle, CB1 et CB2 sont mieux compris comme des nœuds de contrôle intégrés dans des circuits lipidiques, immunitaires et synaptiques. Leur aptitude au développement thérapeutique est réelle. Leur capacité à surprendre l’est tout autant.
CB1 comme cible médicamenteuse : promesses, risques et problème des effets centraux
CB1 demeure l’une des cibles les plus séduisantes en neuropharmacologie, car il occupe un point de contrôle stratégique dans la signalisation synaptique. Les travaux de pharmacologie des récepteurs d’Allyn Howlett ont établi que les effets des cannabinoid n’étaient pas un vague phénomène membranaire, mais une biologie médiée par des récepteurs, et la découverte ultérieure de l’anandamide par Raphael Mechoulam et Lumír Hanuš a doté ce récepteur d’un système de signalisation endogène. Cette histoire est importante. Un récepteur qui contribue à ajuster la libération des neurotransmetteurs à travers les circuits de la douleur, les circuits de l’alimentation, les voies du stress et les réseaux de récompense apparaît comme une cible plausible pour plusieurs troubles difficiles à la fois. Elle crée aussi un piège : lorsqu’un récepteur est intégré dans autant de fonctions cérébrales fondamentales, une modulation large ne reste que rarement confinée à l’effet souhaité.
Ce problème n’est pas théorique. WHO estime que 200 millions de personnes ont utilisé cannabis en 2019, soit environ 4 % de la population mondiale âgée de 15 à 64 ans ; la pharmacologie de CB1 agit donc déjà à l’échelle populationnelle via les cannabinoid de la plante, en particulier Delta-9-THC. Pourtant, les médicaments cannabinoid approuvés restent peu nombreux : FDA indique qu’en 2025 elle avait approuvé un produit médicamenteux dérivé de cannabis et trois produits médicamenteux liés à cannabis. L’écart entre une exposition massive et un nombre limité de thérapeutiques approuvées dit quelque chose d’important sur CB1. La biologie est convaincante. La mise en médicament est difficile.
Fondement thérapeutique dans la douleur, l’appétit et la neuropsychiatrie
L’attrait commence par l’endroit où CB1 agit et par ce qu’il fait. CB1 est fortement exprimé dans de nombreuses terminaisons présynaptiques du système nerveux central, où les endocannabinoid sont souvent produits à la demande dans les cellules postsynaptiques et diffusent rétrogradement à travers la synapse pour supprimer la libération des transmetteurs. Ce système rétrograde peut atténuer le glutamate, le GABA et d’autres flux de signalisation selon le type cellulaire et le circuit. Pour la douleur, il s’agit d’un rationnel évident : si près d’un adulte sur cinq aux États-Unis vit avec une douleur chronique, comme l’ont noté les National Academies en 2017, un récepteur capable de réduire la transmission nociceptive et de modifier l’affect douloureux ne peut qu’attirer l’attention.
Mais CB1 n’est pas un simple interrupteur analgésique. Dans certains circuits, la diminution de la libération inhibitrice du GABA peut désinhiber des neurones en aval plutôt que les apaiser. Dans d’autres, la suppression glutamatergique domine. L’effet dépend de la synapse, de l’état du réseau et du ligand. Les endocannabinoid tels que l’anandamide et le 2-AG sont des signaux brefs, produits localement. Les phytocannabinoid et les agonistes synthétiques arrivent avec des cinétiques différentes, des efficacités différentes et souvent une engagement récepteur beaucoup plus large dans le temps. Cette distinction aide à expliquer pourquoi « l’activation de CB1 réduit la douleur » est trop simpliste pour guider une conception clinique sûre.
L’appétit constitue un deuxième rationnel majeur. La signalisation de CB1 favorise la prise alimentaire et interagit avec les circuits hypothalamiques et mésolimbiques qui relient l’homéostasie énergétique à la récompense. Cela a fait de l’antagonisme de CB1 une stratégie anti-obésité séduisante et de l’agonisme de CB1, ou du renforcement indirect de cette voie, une piste potentielle pour la cachexie ou la perte d’appétit. La logique n’était pas irrationnelle. Elle était fondée sur la physiologie de base. Pourtant, l’appétit est indissociable de l’humeur, de la saillance et du traitement du stress, tous également modulés par CB1.
C’est en neuropsychiatrie que le récepteur devient à la fois le plus intéressant et le plus dangereux. CB1 module les circuits liés à la dopamine, l’inhibition corticale, la plasticité hippocampique, l’apprentissage de la peur et la réactivité au stress. Cela lui confère une pertinence théorique pour les troubles anxieux, les affections liées aux traumatismes, la dépression, les troubles liés à l’usage de substances et les maladies psychotiques. The American Journal of Psychiatry a soutenu en 2025 qu’une signalisation biaisée de CB1 constituait une stratégie plausible pour la schizophrénie, un trouble qui touche environ 24 millions de personnes dans le monde selon WHO. L’importance de cet article ne tient pas au fait qu’un médicament CB1 pour la schizophrénie soit prêt. Elle tient au fait que le domaine est passé d’un agonisme et d’un antagonisme grossiers à une pharmacologie sélective des voies, en s’inspirant du cadre plus large des GPCR.
Pourquoi la cible CB1 est scientifiquement attrayante mais cliniquement difficile
CB1 est scientifiquement attrayant pour exactement la même raison qu’il est cliniquement difficile : c’est un nœud de signalisation dense. Au niveau canonique, CB1 se couple principalement aux protéines Gi/o, réduisant l’activité de l’adénylate cyclase, modulant les canaux ioniques et supprimant la libération des neurotransmetteurs. Cela suffirait déjà à le rendre important. Mais il recrute aussi les beta-arrestins, subit une désensibilisation et une internalisation, et peut être façonné par des modulateurs allostériques et des états conformationnels spécifiques au ligand. La revue structurelle de 2026 dans Frontiers in Chemical Biology le montre clairement pour CB1 comme pour CB2 : la sélectivité et l’efficacité des ligands découlent de différences structurelles au niveau du récepteur qui influencent la liaison, le résultat de signalisation et la régulation du récepteur. Des cannabinoid apparemment similaires peuvent donc produire une pharmacologie significativement différente.
Il ne s’agit pas seulement d’un détail de chimie médicinale. La question touche directement aux effets indésirables. Un ligand qui favorise fortement une conformation active peut induire davantage d’internalisation du récepteur, davantage de tolérance ou un profil comportemental différent de celui d’un ligand stabilisant un autre état. Une étude indexée dans PubMed en 2025/2026 sur la sélectivité des sous-types a en outre montré que la sélectivité des endocannabinoid est dynamique, et non un simple mécanisme verrou-clé, renforçant l’idée que le comportement du récepteur change selon le contexte conformationnel. Les développeurs de médicaments ne font pas face à une seule cible CB1. Ils font face à un ensemble mobile.
C’est pourquoi les agonistes et antagonistes CB1 francs ont si souvent déçu. Un fort agonisme central peut altérer la mémoire, l’attention, les performances psychomotrices et la régulation de l’anxiété, tout en sollicitant les voies de récompense d’une manière qui complique le risque de dépendance. Un antagonisme central fort peut certes réduire l’appétit, mais il peut aussi perturber les mêmes circuits de tamponnement du stress et de régulation affective que les cannabinoid endogènes stabilisent normalement. CB1 participe profondément à la cognition, à la récompense et à la régulation du stress. Tout médicament qui pousse trop fortement ce système dans une seule direction doit être présumé risqué jusqu’à preuve du contraire.
La biologie des systèmes renforce cette prudence. Une analyse de réseau intégrative de 2025/2026 a identifié CB1 et CB2 comme des nœuds hautement influents du système endocannabinoid et a relié la signalisation des récepteurs à des voies métaboliques plus larges. Ce résultat plaide contre une simplification centrée uniquement sur le récepteur. Modifier CB1 ne déplace pas seulement une synapse. Cela peut se répercuter sur des réseaux endocriniens, immunitaires, métaboliques et comportementaux.
Leçons tirées des stratégies à action centrale versus à restriction périphérique
La leçon la plus claire de la première époque du développement des médicaments CB1 est que l’exposition centrale est souvent l’endroit où efficacité et toxicité deviennent indissociables. Les agonistes CB1 à action centrale peuvent engager les circuits de la douleur et de l’appétit, mais la pénétration cérébrale accroît aussi la probabilité d’effets de type intoxication, de sédation, de perturbation cognitive et d’événements indésirables psychiatriques. Les antagonistes CB1 à action centrale illustrent l’échec inverse : ils peuvent atteindre des objectifs métaboliques tout en provoquant des effets inacceptables sur l’humeur et l’anxiété, car ils bloquent aussi le tonus cannabinoid endogène dans les circuits limbiques.
C’est pourquoi les ligands CB1 à restriction périphérique sont devenus attrayants. L’idée est simple : si certains bénéfices thérapeutiques dépendent de CB1 dans des tissus périphériques tels que les afférences primaires, l’intestin, le foie, le tissu adipeux ou d’autres sites non-CNS, alors maintenir un médicament hors du cerveau pourrait préserver les effets utiles tout en réduisant les effets indésirables centraux. Pour la douleur, cela pourrait signifier une action sur des mécanismes nociceptifs périphériques. Pour la maladie métabolique, cela pourrait réduire la lipogenèse ou modifier des voies liées au glucose sans perturber l’humeur ou la cognition.
La logique est solide, mais ce n’est pas une solution miracle. D’abord, la biologie de CB1 périphérique et centrale n’est pas nettement séparée dans les états pathologiques. La douleur chronique, l’alimentation et le stress impliquent tous des boucles cerveau-corps. Ensuite, l’exclusion de la barrière hémato-encéphalique est une question quantitative, pas binaire. De petites quantités de pénétration dans le SNC peuvent encore compter, surtout en cas d’administration chronique. Enfin, un ligand CB1 véritablement périphérique peut malgré tout échouer cliniquement si la maladie ciblée dépend fortement des circuits centraux ou si des voies compensatoires atténuent l’efficacité.
La stratégie plus récente ne consiste donc pas seulement à « rester hors du cerveau ». Elle consiste à « modeler le signal ». Cela inclut les agonistes partiels, les antagonistes neutres plutôt que les agonistes inverses, les modulateurs allostériques et les ligands biaisés favorisant certaines voies de signalisation en aval plutôt que d’autres. La discussion de 2025 dans The American Journal of Psychiatry sur la signalisation biaisée de CB1 dans la schizophrénie s’inscrit dans cette logique. Si un ligand peut préserver certaines voies thérapeutiques tout en réduisant le recrutement des beta-arrestins, la désensibilisation du récepteur ou les effets indésirables sur les circuits, alors CB1 pourrait devenir plus abordable. Peut-être. Rien ne garantit encore que le biais de voie observé dans des systèmes cellulaires se traduira proprement en fenêtres thérapeutiques humaines.
Ainsi, l’état actuel de CB1 comme cible médicamenteuse n’est ni dédaigneux ni romantique. CB1 est attrayant parce qu’il régule le gain synaptique précisément dans les types de circuits que les cliniciens souhaitent influencer. Il est difficile parce que ces circuits gouvernent aussi la mémoire, l’affect, la motivation et la résilience au stress. Les ligands à restriction périphérique, les stratégies de signalisation sélective et une meilleure compréhension structurelle ont amélioré la logique de conception. Ils n’ont pas supprimé le problème fondamental. Un récepteur aussi central dans la fonction cérébrale tolère rarement une pharmacologie brutale.
CB2 comme cible médicamenteuse : neuroinflammation, neurodégénérescence et modulation immunitaire
CB2 est devenu l’une des cibles les plus séduisantes de la pharmacologie du cannabinoid pour une raison simple : il semble offrir un accès à la biologie immunitaire et gliale avec moins de risque d’intoxication, de troubles de la mémoire et de potentiel d’abus classiquement associés à une forte activation de CB1. Cet attrait compte bien au-delà de la cartographie académique des récepteurs. L’Organisation mondiale de la Santé a estimé que 200 millions de personnes avaient utilisé cannabis en 2019, soit 4 % de la population mondiale âgée de 15 à 64 ans, et la FDA américaine ne répertorie encore, en 2025, qu’un seul médicament dérivé de cannabis et trois médicaments liés à cannabis comme approuvés. L’intérêt est élevé. La traduction est difficile.
L’ancienne formule disait que CB1 est le récepteur du cerveau et CB2 le récepteur immunitaire. Aujourd’hui, cette simplification est trop grossière pour être utile. La revue de 2026 publiée dans Frontiers in Behavioral Neuroscience, explicitement présentée comme « une mise à jour des 3 dernières années », considère CB2 comme pertinent pour les troubles du système nerveux central précisément parce qu’il participe à des mécanismes neuroinflammatoires et neurodégénératifs, notamment l’activation microgliale, la signalisation des cytokines et la communication neurone-glie. Le changement clé n’est pas que CB2 soit soudainement devenu abondant dans tout le cerveau. Ce n’est pas le cas. Le changement est qu’une faible expression basale dans de nombreuses régions du SNC ne signifie pas une absence de pertinence fonctionnelle, en particulier lorsque des états pathologiques peuvent modifier l’expression des récepteurs, leur trafic et leur signalisation.
Pourquoi CB2 est souvent considéré comme une cible plus sûre
L’argument de sécurité en faveur de CB2 commence par sa distribution, mais ne s’y limite pas. CB2 est enrichi dans les cellules immunitaires et souvent régulé à la hausse dans la microglie activée, les macrophages infiltrants et d’autres populations cellulaires inflammatoires. À l’inverse, CB1 est fortement exprimé aux terminaisons présynaptiques du cortex, de l’hippocampe, des ganglions de la base et du cervelet, où il module puissamment la libération des neurotransmetteurs. Cette différence anatomique est importante, car un ligand qui engage principalement CB2 est moins susceptible de reproduire le profil psychoactif classique associé à CB1.
Moins susceptible ne signifie pas impossible. La sélectivité est une cible mouvante. La revue de 2026 publiée dans Frontiers in Chemical Biology souligne que la sélectivité « CB1 et CB2 » dépend de différences structurales qui modifient la liaison, l’efficacité et la régulation du récepteur, et une étude récente indexée dans PubMed sur la sélectivité de sous-type soutient que la discrimination des endocannabinoids entre sous-types de récepteurs reflète une dynamique conformationnelle plutôt qu’un modèle fixe de type clé-serrure. En pratique, cela signifie qu’un composé peut sembler sélectif de CB2 dans un test et se comporter de manière moins nette dans un autre, en particulier lorsque la densité des récepteurs, l’environnement membranaire et les effecteurs en aval diffèrent. Le même ligand peut biaiser la signalisation par protéines G dans un type cellulaire, recruter des beta-arrestins dans un autre, et présenter des cinétiques de désensibilisation ou d’internalisation différentes en conditions inflammatoires.
Cette complexité mécanistique renforce en réalité l’intérêt d’explorer CB2 avec prudence. Si l’objectif thérapeutique est de réduire la production de cytokines par la microglie, de limiter le recrutement des cellules immunitaires périphériques ou de modifier le stress métabolique glial sans perturber fortement la transmission synaptique corticale, alors CB2 demeure le premier récepteur le plus plausible à cibler. La signalisation biaisée de CB1, comme l’argumente l’article de 2025 du American Journal of Psychiatry sur la schizophrénie, pourrait à terme élargir la fenêtre thérapeutique des médicaments dirigés contre CB1. Mais cela constitue un argument en faveur d’une pharmacologie plus intelligente de CB1, non une raison d’ignorer la moindre charge psychoactive des approches centrées sur CB2.
Plus sûr, toutefois, ne doit pas être confondu avec efficace. Un médicament peut éviter une intoxication manifeste tout en échouant parce que le récepteur est exprimé trop faiblement, trop variablement ou trop brièvement au cours du processus pathologique pour produire un effet clinique significatif. Cela s’est produit à de nombreuses reprises dans le développement de médicaments contre la neuroinflammation, et pas seulement dans la science du cannabis.
Preuves et limites dans les modèles de maladies du SNC
La revue de Frontiers in Behavioral Neuroscience explique pourquoi CB2 réapparaît sans cesse dans les modèles de maladie d’Alzheimer, de maladie de Parkinson, de sclérose en plaques, de traumatisme crânien, d’AVC et de douleur neuropathique : ces affections comportent toutes une signalisation inflammatoire, une réactivité gliale, un stress oxydatif et des dommages tissulaires progressifs. Dans de nombreuses études chez le rongeur, les agonistes de CB2 réduisent des médiateurs pro-inflammatoires tels que TNF-α, IL-1β et IL-6, orientent la microglie vers des phénotypes moins délétères ou atténuent l’infiltration leucocytaire. Cela s’accompagne parfois d’une amélioration du comportement moteur, d’une réduction de la taille des lésions ou d’une préservation des marqueurs synaptiques. Ce sont des signaux réels, et non des artefacts à écarter.
Mais les résultats sont inégaux. Les effets dépendent souvent du moment d’administration. Un agoniste de CB2 administré pendant la phase inflammatoire précoce après une lésion peut agir très différemment du même agoniste administré au cours d’une dégénérescence chronique. La dose compte aussi, tout comme la classe du ligand. Les endocannabinoids tels que 2-AG et l’anandamide ne sont pas interchangeables avec les phytocannabinoids ou avec des agonistes synthétiques préférentiels de CB2. Leurs concentrations locales, leur dégradation métabolique et leurs actions hors cible diffèrent. Il en va de même pour leurs préférences de voie.
Les modèles de maladie eux-mêmes constituent une autre limite. Une souris transgénique Alzheimer présentant une pathologie amyloïde et une microglie réactive n’est pas la maladie d’Alzheimer chez un être humain de 74 ans présentant une défaillance mixte vasculaire, inflammatoire et protéostatique. L’encéphalomyélite auto-immune expérimentale est utile pour la biologie de la sclérose en plaques, mais elle n’en capture qu’une partie. Les résultats précliniques positifs établissent donc une plausibilité, pas une preuve.
Il existe aussi un problème persistant de mesure en biologie de CB2. La spécificité des anticorps a longtemps constitué un point faible, rendant les affirmations de localisation du récepteur plus difficiles à croire que ne le laissent entendre de nombreux articles. Les faibles niveaux d’expression dans le cerveau sain, l’expression inductible dans la maladie et les différences entre espèces compliquent encore l’interprétation. Certaines observations rapportées de « CB2 neuronal » peuvent être réelles dans des contextes spécifiques, mais les affirmations larges d’une expression neuronale constitutive et étendue de CB2 restent moins solides que le champ ne l’a longtemps suggéré.
C’est ici que la signalisation importe autant que l’anatomie. CB2 est un GPCR couplé à Gi/o, mais ce raccourci masque d’importantes variations : l’inhibition de l’adénylate cyclase, la modulation des voies MAPK, les effets sur les canaux ioniques, le recrutement de beta-arrestins, la désensibilisation du récepteur et l’internalisation peuvent tous modifier le résultat biologique net. Un ligand qui réduit la libération de cytokines dans des microglies en culture peut échouer in vivo s’il désensibilise rapidement les récepteurs ou s’il déclenche un programme dominé par les arrestines peu favorable. Une moindre propension à l’intoxication ne résout pas ces problèmes pharmacologiques.
Ce que la littérature récente suggère pour les thérapies futures
La littérature récente s’éloigne de l’ancienne recherche d’un agoniste unique de CB2 pour l’inflammation cérébrale et s’oriente vers une stratégie plus sélective et dépendante du contexte. La revue structurale de 2026 et l’article de 2025/2026 sur la sélectivité des sous-types soutiennent toutes deux la même leçon : la cible thérapeutique dépendra de l’état conformationnel, du profil d’efficacité et du contexte tissulaire, et pas seulement de l’affinité nominale pour un sous-type. La conception des médicaments se dirige vers des ligands sélectifs sur le plan fonctionnel, et pas seulement sur le plan de la liaison.
Cela signifie probablement trois choses. Premièrement, les futurs médicaments CB2 devront présenter des empreintes de signalisation plus fines, incluant le biais entre protéines G et beta-arrestin, le temps de résidence sur le récepteur et les effets spécifiques au type cellulaire dans des systèmes humains plutôt que dans de simples lignées immortalisées. Deuxièmement, la stratification des maladies sera essentielle. Une thérapie visant CB2 pourrait être plus efficace lorsque l’amplification inflammatoire est un moteur majeur, et non un simple sous-produit, de la pathologie. Troisièmement, une approche combinatoire est inévitable. Une analyse intégrative de réseau publiée dans une étude récente indexée dans PubMed a identifié CB1 et CB2 comme des nœuds influents dans des voies plus larges liées à endocannabinoid et au métabolisme, ce qui correspond à la réalité clinique des maladies du SNC : inflammation, stress mitochondrial, signalisation lipidique et dysfonction synaptique sont intimement imbriqués.
Dans certaines conditions inflammatoires, la modulation de CB2 peut réduire la signalisation immunitaire délétère tout en évitant une grande partie de la charge psychoactive liée à CB1.Preliminary evidence
Cela ne fait pas de CB2 une impasse. Cela en fait une cible qui relève probablement de la pharmacologie de précision plutôt que de la mythologie réceptrice. L’argument actuel le plus solide n’est pas « l’activation de CB2 traite la neurodégénérescence ». Il est plus étroit : dans certaines conditions inflammatoires, dans certaines populations cellulaires, avec les bonnes propriétés de ligand, la modulation de CB2 peut réduire une signalisation immunitaire délétère tout en évitant une grande partie de la charge psychoactive liée à CB1. C’est une hypothèse thérapeutique crédible. Ce n’est pas encore une règle cliniquement validée.
Les composés du cannabis sur CB1 et CB2 : THC, CBD, cannabinoïdes mineurs et ligands synthétiques
Le cannabis n’agit pas par l’intermédiaire d’une seule substance chimique ni d’un seul mécanisme récepteur. Cela paraît évident, pourtant le débat public réduit encore souvent la pharmacologie à une fausse équation : THC=CB1, CBD=CB2. La littérature ne valide pas ce raccourci. Le cannabis contient de nombreux phytocannabinoïdes, ainsi que des terpènes, des flavonoïdes et d’autres constituants, tandis que l’organisme produit ses propres endocannabinoids, tels que l’anandamide et le 2-arachidonoylglycérol, identifiés grâce à des travaux associés à Raphael Mechoulam, Lumír Hanuš et d’autres. De plus, la majeure partie des connaissances scientifiques sur l’activation des récepteurs, l’efficacité, la sélectivité, la désensibilisation et le biais de signalisation provient de ligands synthétiques conçus comme sondes ou comme pistes médicamenteuses, plutôt que des seuls composés végétaux.
Cette distinction est importante, car CB1 et CB2 ne sont pas des verrous passifs attendant une clé cannabinoid. Ce sont des GPCR de classe A présentant plusieurs états conformationnels, des préférences de couplage et des devenirs régulateurs différents. Une revue structurale de 2026 dans Frontiers in Chemical Biology le formule clairement : la sélectivité des ligands pour CB1 et CB2 dépend de différences structurales au niveau des récepteurs qui modifient la liaison, l’efficacité et la régulation du récepteur, et pas seulement du fait qu’une molécule « touche » l’un ou l’autre récepteur. Une étude récente indexée dans PubMed sur la sélectivité de sous-type soutient également que la préférence des endocannabinoids pour CB1 ou CB2 découle de dynamiques conformationnelles plutôt que d’un simple ajustement clé-serrure. Ainsi, lorsque deux cannabinoïdes paraissent chimiquement similaires mais produisent des effets différents dans le cerveau, les tissus immunitaires ou les circuits de la douleur, ce n’est pas un détail secondaire. C’est la pharmacologie fondamentale.
Étant donné que l’Organisation mondiale de la santé a estimé que 200 millions de personnes ont consommé du cannabis en 2019, soit environ 4 % de la population mondiale âgée de 15 à 64 ans, ces distinctions ne relèvent pas de la trivia académique. Elles déterminent l’intoxication, les allégations thérapeutiques, la sécurité, et expliquent pourquoi certains médicaments ciblant des récepteurs réussissent tandis que d’autres échouent.
Comment le THC engage CB1 et contribue aux effets psychoactifs
Le Delta-9-tetrahydrocannabinol, ou THC, reste le composé central pour comprendre l’intoxication liée au cannabis, car il engage directement CB1 dans le système nerveux central. C’est l’explication la plus nette, au niveau du récepteur, des effets psychoactifs caractéristiques du cannabis, même si le terme « psychoactif » recouvre plusieurs effets distincts : altération de la perception du temps, de la récompense, perturbation de la mémoire, anxiogénèse chez certains utilisateurs et altération de la coordination motrice. Les travaux de pharmacologie des récepteurs menés par Allyn Howlett ont contribué à établir ce cadre centré sur CB1, et il demeure fondamental.
Mais « le THC active CB1 » n’est que le point de départ. Le THC n’est pas un interrupteur parfait. Il est généralement décrit comme un agoniste partiel de CB1, ce qui signifie que son efficacité dépend de la densité des récepteurs, du contexte tissulaire et du test de signalisation utilisé. Dans des régions riches en CB1 comme le cortex, l’hippocampe, les ganglions de la base et le cervelet, cela peut suffire à produire des effets centraux marqués. Au niveau synaptique, CB1 est souvent présynaptique, où son activation supprime la libération des neurotransmetteurs. Cela signifie que le THC peut atténuer la libération de glutamate dans un circuit, la libération de GABA dans un autre, et produire des effets opposés au niveau du réseau selon les cellules exprimant le récepteur et le moment considéré.
C’est pourquoi la localisation du récepteur ne peut pas être réduite à « CB1 dans le cerveau ». CB1 est abondant dans le cerveau, certes, mais il n’est ni uniformément réparti, ni statique, ni fonctionnellement identique selon les types de neurones. Il en résulte que les effets du THC dépendent des circuits. L’euphorie et le renforcement font partie du tableau. Il en va de même de la perturbation de la mémoire à court terme et des signalisations pertinentes pour la psychose chez les personnes vulnérables.
Ce dernier point est devenu plus important, et non moins. Un article de 2025 dans l’American Journal of Psychiatry a soutenu que la signalisation biaisée de CB1 constitue une stratégie thérapeutique plausible dans la schizophrénie, précisément parce que la biologie de CB1 ne peut pas être comprise comme une simple activation récepteur unidimensionnelle. La schizophrénie touche environ 24 millions de personnes dans le monde selon l’WHO, et toute discussion sérieuse sur le THC au niveau de CB1 doit tenir compte du fait que la même famille de récepteurs impliquée dans l’intoxication est aussi étudiée à travers l’agonisme biaisé, le recrutement de beta-arrestin et la conception de médicaments sélectifs de voie. Le THC peut engager une signalisation couplée à Gi/o, mais CB1 subit également phosphorylation, désensibilisation et internalisation, avec des conséquences sur la tolérance et la réponse en aval. La dose et le moment d’exposition comptent. Les expositions répétées modifient le système.
Les ligands synthétiques de CB1 l’ont montré depuis longtemps. Des composés tels que CP55,940 et WIN55,212-2 sont de puissants agonistes de recherche qui produisent souvent, dans les essais de laboratoire, des réponses réceptrices plus fortes ou plus nettes que le THC. Le rimonabant, inverse agoniste de CB1, a montré la direction opposée : bloquer CB1 ou l’abaisser sous son activité basale pouvait réduire l’appétit et le poids corporel, mais les effets indésirables psychiatriques ont entraîné l’échec du médicament. Cette histoire clinique est un avertissement. Le ciblage sélectif de CB1 peut être pharmacologiquement élégant tout en restant médicalement décevant.
Pourquoi le CBD ne correspond pas à une histoire simple d’agoniste CB1/CB2
Le CBD est le cannabinoid le plus souvent réduit à un mythe simplificateur. Il est couramment présenté comme « celui qui n’est pas intoxicant et agit via CB2 » ou comme l’opposé direct du THC au niveau du récepteur. Aucune de ces affirmations n’est exacte.
| Classe de composé ou exemple | Comment l’article décrit le comportement du récepteur | Conclusion essentielle |
|---|---|---|
| THC | Agoniste partiel de CB1 et CB2 ; central dans l’intoxication via CB1 | L’engagement de CB1 aide à expliquer les effets psychoactifs, mais l’issue reste contextuelle |
| CBD | Pas un agoniste direct simple de CB1 ou CB2 ; peut agir de manière allostérique ou indirecte | Le CBD n’est pas un simple miroir de THC pour CB2 |
| Cannabinoïdes mineurs | Les effets signalés varient selon le test et la concentration | Ne pas généraliser le comportement récepteur d’un cannabinoïde à tous |
| Ligands synthétiques | Souvent utilisés pour définir l’efficacité, la sélectivité et le biais | Les composés sondes peuvent se comporter très différemment des cannabinoïdes végétaux |
Le CBD ne se comporte pas comme un agoniste CB1 ou CB2 direct de la même manière que le THC agit sur CB1. Son affinité pour les sites orthostériques de CB1 et CB2 est relativement faible, et une grande partie de sa pharmacologie semble impliquer des actions indirectes, allostériques ou hors des récepteurs cannabinoid. Selon le système étudié, il a été rapporté que le CBD agit comme modulateur allostérique négatif de CB1, ce qui pourrait modifier la manière dont le THC ou les endocannabinoids signalent sans simplement activer ou désactiver le récepteur. Cette distinction est cruciale. Moduler la forme du récepteur et l’efficacité de la signalisation n’est pas la même chose que l’agonisme classique.
Le CBD interagit également avec des cibles au-delà de CB1 et CB2, notamment TRPV1, 5-HT1A, PPAR-gamma, des voies liées à l’adénosine et des enzymes impliquées dans le tonus endocannabinoid. Certains effets peuvent refléter une signalisation de l’anandamide modifiée plutôt qu’une stimulation directe du récepteur. Cela aide à expliquer pourquoi le profil clinique du CBD ne s’aligne pas nettement sur l’ancien raccourci récepteur. La FDA indique avoir approuvé, en 2025, un produit médicamenteux dérivé du cannabis et trois produits médicamenteux liés au cannabis ; l’usage médical le mieux établi du CBD se situe dans l’épilepsie, une affection touchant environ 50 millions de personnes dans le monde, et cette voie thérapeutique ne peut pas être expliquée par un modèle caricatural selon lequel « le CBD se lie à CB2 et réduit l’inflammation ».
Même pour CB2, la situation est plus large que l’ancienne narration périphérique. Une revue de 2026 dans Frontiers in Behavioral Neuroscience a décrit « une mise à jour au cours des 3 dernières années » dans laquelle la signalisation de CB2 a gagné en attention dans les troubles du système nerveux central, en lien avec des mécanismes neuroinflammatoires et neurodégénératifs. Cela ne signifie pas que le CBD soit simplement un médicament de CB2. Cela signifie que le récepteur lui-même a une portée fonctionnelle plus large que ne le suggère le récit public.
L’importance de distinguer les composés végétaux des mythes récepteurs
Les phytocannabinoïdes ne sont pas interchangeables, et ils ne sont pas interchangeables avec les ligands synthétiques. Les cannabinoïdes mineurs tels que le cannabigerol (CBG), le cannabinol (CBN), le tetrahydrocannabivarin (THCV) et le cannabichromene (CBC) ont été décrits comme interagissant avec CB1 et CB2 de manière variable selon le test et la concentration. Certains montrent une faible agonisme, d’autres une agonisme partiel, d’autres encore une antagonisme dans certaines conditions, et certains présentent une activité significative sur des cibles non cannabinoid. Le THCV en est un bon exemple : il a été décrit comme antagoniste de CB1 ou antagoniste neutre à faibles doses dans certains systèmes et comme agoniste partiel à des doses plus élevées dans d’autres. Il ne s’agit pas d’une incohérence gratuite. Cela reflète une pharmacologie récepteur dépendante du contexte.
Les outils de recherche précisent ce point. Les endocannabinoids, les phytocannabinoïdes et les ligands synthétiques peuvent tous activer « CB1 et CB2 », mais ils ne stabilisent pas les mêmes conformations réceptrices et ne produisent pas le même biais à travers les protéines G et les beta-arrestins. La revue structurale de 2026 et les analyses récentes de réseau indexées dans PubMed orientent toutes deux le domaine loin de la mythologie réceptrice et vers une pharmacologie des systèmes. Les récepteurs s’inscrivent dans des réseaux de signalisation spécifiques à chaque cellule, des voies métaboliques et des mécanismes de trafic intracellulaire. Le profil clinique d’un ligand émerge de cet ensemble, et non d’une étiquette telle que « CB2 anti-inflammatoire » ou « CBD équilibre le THC ».
Pour un wiki sur le cannabis, c’est là que réside la ligne la plus importante : le THC a une pertinence principale pour l’intoxication médiée par CB1, mais la pharmacologie du cannabis ne s’arrête pas là, et le CBD n’est pas une simple image miroir de THC sur CB2. Les composés végétaux ouvrent l’histoire. L’état du récepteur, la distribution tissulaire, l’efficacité, le biais et la régulation décident de la manière dont elle se termine.
Schizophrénie, risque de psychose, et ce que la biologie de CB1 implique et n’implique pas
Pourquoi la schizophrénie entre dans la conversation sur les récepteurs
La schizophrénie est importante ici parce que CB1 n’est pas simplement un récepteur de plus sur une longue liste de cibles. C’est l’un des récepteurs couplés aux protéines G les plus abondants dans le cerveau, situé sur les terminaisons présynaptiques où il peut atténuer la libération des neurotransmetteurs et remodeler l’activité des réseaux dans des circuits déjà impliqués dans la psychose : cortex, hippocampe, striatum, amygdale. Lorsqu’un récepteur se trouve aussi près du contrôle circuitaire du glutamate, du GABA, de la dopamine et de la réactivité au stress, les chercheurs en psychiatrie y prêtent attention.
L’échelle de santé publique en fait davantage qu’une question académique. L’Organisation mondiale de la Santé estime que le cannabis a été consommé par 200 millions de personnes en 2019, soit 4 % de la population mondiale âgée de 15 à 64 ans. L’OMS estime également que la schizophrénie touche environ 24 millions de personnes dans le monde, soit à peu près 1 personne sur 300. Ce ne sont pas de petites populations isolées. Elles se recoupent dans les services cliniques, aux urgences et dans les études de cohorte longitudinales. Toute biologie des récepteurs susceptible d’éclairer le risque de psychose, le développement de médicaments antipsychotiques, ou les deux, mérite un examen sérieux.
C’est ce contexte translationnel qui sous-tend l’article de 2025 du American Journal of Psychiatry avançant que le signalement biaisé de CB1 constitue une stratégie thérapeutique plausible pour la schizophrénie. L’idée centrale est facile à formuler et facile à déformer. Facile à formuler : CB1 ne se comporte pas comme un simple interrupteur marche/arrêt, et différents ligands peuvent stabiliser différentes conformations du récepteur, orientant la signalisation vers certaines voies intracellulaires ou à l’écart de celles-ci. Facile à déformer : si CB1 est pertinent pour la recherche sur le traitement de la schizophrénie, certains lecteurs en déduisent à tort que « l’activation cannabinoid aide la schizophrénie » ou que la consommation de cannabis est donc, par défaut, médicinale. L’article ne soutient pas ce saut logique.
Historiquement, la science des cannabinoid est restée prisonnière de raccourcis. CB1 a été présenté comme le « récepteur cérébral », CB2 comme le « récepteur immunitaire », et les effets psychoactifs ont été attribués presque entièrement à l’occupation de CB1. Cette représentation a toujours été incomplète et, en 2025, elle est clairement trop rudimentaire. Les travaux fondateurs d’Allyn Howlett ont établi la pharmacologie des récepteurs cannabinoid, tandis que Mechoulam et Hanus ont contribué à définir la biologie des endocannabinoid à travers l’anandamide. Depuis lors, la pharmacologie des récepteurs est passée de la présence du récepteur à son état fonctionnel. Ce changement compte pour la schizophrénie, car la physiopathologie n’est pas causée par la seule expression du récepteur. Elle émerge du timing, du type cellulaire, du contexte circuitaire et de la réponse intracellulaire.
La littérature plus récente renforce ce point. Une revue de 2026 dans Frontiers in Chemical Biology explique que les différences structurales entre CB1 et CB2 façonnent la liaison des ligands, leur efficacité, leur signalisation et la régulation des récepteurs. Une étude indexée dans PubMed de 2025/2026 sur la sélectivité des sous-types soutient que la sélectivité des endocannabinoid peut découler de dynamiques conformationnelles plutôt que d’un modèle statique de clé et serrure. Un autre article d’analyse de réseaux de 2025/2026 identifie CB1 et CB2 comme des nœuds hautement influents de l’ensemble du système endocannabinoid, reliés à des voies métaboliques et de signalisation plutôt qu’à des boîtes réceptrices isolées. Pour la recherche sur la schizophrénie, cela signifie qu’il faut cesser de demander seulement « la molécule cible-t-elle CB1 ? » et commencer à demander « quels états de CB1, dans quelles cellules, dans quelles conditions d’exposition ? »
C’est là que réside le sérieux clinique. Pas dans des formules simplistes sur les récepteurs. Dans la précision mécanistique.
Signalisation biaisée versus activation brutale du récepteur
L’activation brute de CB1 est un mauvais modèle pour raisonner en termes thérapeutiques. Elle réduit trop de variables à un seul mot : activation. Les ligands endogènes comme l’anandamide et le 2-AG, les phytocannabinoid comme delta-9-THC, et les agonistes synthétiques ne se comportent pas de manière identique sur CB1, même s’ils comptent tous comme ligands. Ils peuvent différer par l’affinité, l’efficacité, le temps de résidence, l’exposition régionale et le biais de voie. Ils peuvent aussi différer quant à leur capacité à recruter les protéines Gi/o par rapport aux beta-arrestines, à déclencher une désensibilisation, et à induire une internalisation du récepteur après des doses répétées.
Cette distinction est centrale dans l’argument du AJP sur la schizophrénie. Un ligand qui oriente légèrement CB1 vers un programme intracellulaire peut ne pas ressembler à un ligand qui entraîne une activation large du récepteur dans de nombreuses régions cérébrales. En pharmacologie des GPCR, cette différence peut séparer un médicament utile d’un médicament intolérable. CB1 est particulièrement sensible à ce problème, car un même récepteur peut réduire la transmission excitatrice à une synapse, supprimer la transmission inhibitrice à une autre, et modifier la plasticité en aval de manière qui n’est pas fonctionnellement équivalente. De petites différences moléculaires peuvent produire de grandes conséquences à l’échelle des systèmes.
THC est l’exemple évident de la raison pour laquelle « cible CB1 » n’équivaut pas à « thérapie CB1 ». THC est un phytocannabinoid doté de propriétés d’agoniste partiel sur CB1, mais les effets vécus et cliniques de THC ne sont pas une lecture pure d’une seule branche de signalisation. La dose compte. L’âge au moment de l’exposition compte. L’exposition répétée compte. Le rapport entre THC et les autres constituants compte également, tout comme la vulnérabilité psychiatrique de la personne. Des effets aigus de type intoxication, l’anxiété, des modifications perceptives et des symptômes psychotiques transitoires chez certains usagers ne démontrent pas que tous les thérapeutiques ciblant CB1 sont voués à l’échec. Ils montrent en revanche qu’une stimulation indifférenciée de CB1 n’est pas un raccourci vers un traitement antipsychotique.
L’agonisme biaisé est attrayant précisément parce qu’il cherche à éviter cette brutalité. L’espoir thérapeutique n’est pas « plus de CB1 ». C’est « le bon profil de signalisation de CB1 ». Cela peut signifier favoriser les voies liées à la stabilisation des circuits tout en limitant celles associées aux effets cognitifs ou psychotomimétiques indésirables, à la tolérance ou à la diminution de la régulation des récepteurs. Reste à savoir si cela peut être obtenu chez les patients. Le concept est plausible. Ce n’est pas un succès clinique démontré.
CB2 a également sa place dans ce tableau, mais pas comme solution simpliste. La revue de 2026 dans Frontiers in Behavioral Neuroscience décrit « une mise à jour sur les 3 dernières années » dans laquelle le signalement de CB2 a retenu l’attention dans les troubles du système nerveux central en raison de liens neuroinflammatoires et neurodégénératifs. Cela affaiblit l’ancienne affirmation selon laquelle CB2 serait sans pertinence pour les maladies cérébrales. Mais cela ne rétablit pas l’ancien binaire en le renversant. La schizophrénie n’est pas seulement une inflammation, et CB2 n’est pas un simple bouton anti-inflammatoire. Tout modèle translationnel sérieux doit prendre en compte à la fois les interactions neurones-glie, la signalisation immunitaire et les dysfonctionnements des circuits.
Ce qu’il ne faut pas inférer
- Pas une preuve de bénéfice Le fait que CB1 soit une cible médicamenteuse ne signifie pas que l’usage du cannabis protège contre la schizophrénie ou la psychose.
- Pas une réfutation du risque Les études mécanistiques sur les récepteurs n’annulent pas les inquiétudes épidémiologiques liées à une exposition élevée au THC chez les personnes vulnérables.
- Pas d’interchangeabilité Les endocannabinoïdes, les phytocannabinoïdes et les ligands synthétiques ne doivent pas être considérés comme le même type d’exposition.
- Pas d’approbation générale Les autorisations actuelles de la FDA ne tranchent pas les questions de sécurité psychiatrique ni d’efficacité pour l’usage étendu des cannabinoïdes.
Ce que les lecteurs ne doivent pas déduire des études mécanistiques
Premièrement, les lecteurs ne doivent pas en conclure que, parce que CB1 est une cible thérapeutique, la consommation de cannabis est donc protectrice contre la schizophrénie ou la psychose. La logique récepteur-médicament ne fonctionne pas ainsi. Les récepteurs beta sont des cibles thérapeutiques ; cela ne signifie pas que tout stimulant est un traitement. Les récepteurs opioïdes sont des cibles thérapeutiques ; cela ne signifie pas que tout agonisme opioïde est bénéfique. Le même principe s’applique ici.
Deuxièmement, les études mécanistiques n’annulent pas les inquiétudes épidémiologiques. La recherche sur le biais des récepteurs et la conception des ligands coexiste avec les données montrant que l’exposition au cannabis peut aggraver le risque de psychose chez certaines personnes, en particulier avec des produits à forte teneur en THC, un début précoce ou une consommation intensive. Un mécanisme récepteur peut expliquer pourquoi certains composés pourraient être conçus pour de meilleurs résultats. Il ne justifie pas des affirmations publiques générales selon lesquelles « l’activation de CB1 est bonne pour la schizophrénie ».
Troisièmement, les lecteurs ne doivent pas traiter les endocannabinoid, les phytocannabinoid et les ligands synthétiques comme interchangeables. L’anandamide n’est pas THC. Un modulateur de CB1 à restriction périphérique n’est pas du cannabis fumé. Un composé expérimental à faible efficacité et au profil de signalisation biaisé n’est pas un produit de consommation à forte exposition. Ces catégories comptent parce que le comportement du récepteur dépend de la classe du ligand, de la dose, de la voie d’administration, du moment et de l’exposition tissulaire.
Enfin, l’existence de médicaments liés au cannabis déjà approuvés ne tranche pas les questions psychiatriques. En 2025, la FDA note qu’un médicament dérivé du cannabis et trois médicaments liés au cannabis ont été approuvés. Ce fait montre que la pharmacologie des cannabinoid peut devenir un médicament dans des conditions définies. Il ne signifie pas qu’une stimulation non raffinée des récepteurs est sûre, efficace ou appropriée pour la schizophrénie.
La conclusion correcte est plus stricte et moins rassurante. La biologie de CB1 est pertinente pour la psychose parce que le récepteur est profondément intégré dans les circuits cérébraux qui façonnent la saillance, la cognition et l’inhibition. Cette pertinence soutient une conception prudente des médicaments, et non une extrapolation hâtive. Dans la recherche sur la schizophrénie, la pharmacologie des récepteurs est à la fois une carte des possibilités et des risques.
Ce que la science des récepteurs signifie pour les futurs médicaments cannabinoid
La médecine cannabinoid s’éloigne de l’ancienne idée selon laquelle un médicament utile doit simplement « cibler CB1 » ou « cibler CB2 ». Ce raccourci a toujours été incomplet. Il est désormais activement trompeur. CB1 et CB2 ne sont pas de simples interrupteurs marche/arrêt séparés proprement entre cerveau et système immunitaire. Ce sont des récepteurs dotés de distributions chevauchantes mais inégales entre les types cellulaires, de conformations dynamiques, de préférences d’accouplement distinctes et de rôles changeants selon les états pathologiques. Cela importe parce qu’un même ligand peut paraître thérapeutique dans un tissu, intoxicant dans un autre, et inefficace dans un essai clinique si la population malade est trop hétérogène ou si le schéma posologique entraîne une désensibilisation.
Les enjeux de santé publique ne sont pas mineurs. L’Organisation mondiale de la Santé a estimé que 200 millions de personnes ont utilisé cannabis en 2019, soit 4 % de la population mondiale âgée de 15 à 64 ans. Pourtant, la FDA américaine note encore qu’un seul médicament dérivé du cannabis et trois médicaments liés au cannabis ont été approuvés en 2025. Cet écart entre une exposition massive et un nombre limité de thérapeutiques approuvées est l’une des raisons pour lesquelles la pharmacologie des récepteurs compte autant. Une autre raison est le fardeau des maladies : l’épilepsie touche environ 50 millions de personnes dans le monde, la schizophrénie environ 24 millions, et la douleur chronique affecte près d’un adulte sur cinq aux États-Unis. Si la biologie des cannabinoid doit déboucher sur de meilleurs médicaments, cela ne se fera pas par une activation grossière des récepteurs.
Les travaux fondateurs de Raphael Mechoulam, Lumír Hanuš et Allyn Howlett ont établi le domaine moderne en identifiant la signalisation endocannabinoid et la pharmacologie des récepteurs. L’étape suivante est différente. Il s’agit moins de prouver l’existence de CB1 et CB2 que d’apprendre à les orienter avec précision.
| Concept de conception | Ce qu’il vise à améliorer | Pourquoi l’article dit que cela compte |
|---|---|---|
| Sélectivité de sous-type | Préférer plus nettement CB1 ou CB2 | Le sous-type seul ne garantit ni efficacité ni sécurité |
| Ajustement de l’efficacité | Éviter une activation trop forte du récepteur | Différents états actifs peuvent modifier le bénéfice, la tolérance et les effets indésirables |
| Signalisation biaisée | Favoriser une voie intracellulaire plutôt qu’une autre | Peut aider à séparer les effets utiles des inconvénients |
| Modulation allostérique | Ajuster la réponse du récepteur aux ligands endogènes | Pourrait préserver une signalisation spatiale et temporelle plus normale |
| Restriction tissulaire | Limiter l’exposition à certains organes ou éviter la pénétration cérébrale | Utile lorsque les effets indésirables centraux dominent |
Sélectivité, efficacité, modulation allostérique et biais
La leçon la plus importante de la science récente des récepteurs est que « liaison » n’est pas synonyme « d’effet ». Un ligand peut préférer CB1 à CB2, ou inversement, mais cela n’est qu’un début. Il peut aussi différer en efficacité, c’est-à-dire dans sa capacité à stabiliser les états actifs du récepteur. Il peut favoriser la signalisation Gi/o plutôt que le recrutement de beta-arrestin, ou promouvoir davantage l’internalisation du récepteur qu’une signalisation durable à la surface cellulaire. Ces différences peuvent modifier radicalement les résultats.
La revue de 2026 de Frontiers in Chemical Biology sur la structure des récepteurs cannabinoid le montre clairement : la sélectivité de sous-type entre CB1 et CB2 dépend de différences structurelles au niveau du récepteur qui façonnent la liaison du ligand, l’efficacité de la signalisation et la régulation du récepteur. C’est pourquoi des cannabinoid chimiquement proches ne se comportent pas de manière identique. Un phytocannabinoid, un endocannabinoid comme l’anandamide et un ligand synthétique peuvent tous entrer en contact avec la même famille de récepteurs tout en produisant des profils en aval différents. La conception de médicaments qui ignore l’efficacité et le biais de signalisation est désormais en retard sur les données.
CB1 est le cas le plus clair. On le présente souvent comme le récepteur responsable des effets psychoactifs, ce qui est vrai mais incomplet. CB1 régule également la transmission synaptique, le traitement de la douleur, l’appétit, les circuits du stress et la signalisation corticale. L’article de 2025 du American Journal of Psychiatry soutient qu’une signalisation CB1 biaisée constitue une stratégie thérapeutique plausible pour la schizophrénie. C’est un changement remarquable dans le domaine. La schizophrénie touche environ 24 millions de personnes dans le monde, et l’article ne considère pas CB1 comme un récepteur à éviter entièrement. Il le considère plutôt comme une cible dont les conséquences pathologiques et thérapeutiques peuvent dépendre des voies intracellulaires privilégiées par un ligand. C’est de la pharmacologie classique des GPCR, désormais appliquée sérieusement aux thérapeutiques cannabinoid.
CB2 a connu une réévaluation similaire. La revue de 2026 de Frontiers in Behavioral Neuroscience décrit « une mise à jour sur les 3 dernières années » montrant un intérêt croissant pour la signalisation CB2 dans les troubles du système nerveux central, en particulier lorsque des mécanismes neuroinflammatoires et neurodégénératifs sont impliqués. Cela ne signifie pas que CB2 soit une cible anti-inflammatoire magique. Cela signifie que l’ancienne affirmation selon laquelle CB2 est sans pertinence pour le cerveau ne correspond plus à la littérature. L’expression peut être faible dans des conditions de base et augmenter dans certaines populations cellulaires ou dans des contextes pathologiques. Un médicament ciblant CB2 peut donc dépendre fortement du moment, de la pathologie et de l’état du tissu.
La modulation allostérique pourrait devenir l’une des façons les plus utiles d’exploiter ces distinctions. Les agonistes orthostériques entraînent souvent une activation large du récepteur, ce qui augmente le risque d’effets indésirables, de tolérance et de faible sélectivité contextuelle. Les modulateurs allostériques, à l’inverse, peuvent ajuster la réponse des récepteurs aux ligands endogènes plutôt que de forcer une activation maximale. En principe, cela pourrait préserver les aspects spatiaux et temporels de la signalisation endocannabinoid normale. C’est une stratégie séduisante pour CB1, où l’agonisme direct se heurte souvent à des effets indésirables centraux, et pour CB2, où une surexpression liée à la maladie peut rendre une modulation dépendante du contexte plus attrayante qu’une stimulation constante. Mais « séduisante » ne signifie pas « prouvée ». De nombreuses idées allostériques qui paraissent élégantes in vitro échouent encore lorsque la réserve de récepteurs, l’hétérogénéité tissulaire et la pharmacocinétique entrent en jeu.
Pourquoi la biologie structurale et la biologie des réseaux guident désormais la conception des médicaments
Le centre de gravité du domaine s’est déplacé du simple cartographie des récepteurs vers une analyse dynamique des états du récepteur. C’est là que la biologie structurale devient essentielle. La revue de 2026 de Frontiers et l’étude récente indexée dans PubMed sur le mécanisme dynamique de la sélectivité de sous-type s’opposent toutes deux au modèle clé-serrure. La sélectivité n’est pas seulement une question de compatibilité plus ou moins bonne entre un ligand et une poche de récepteur. Les récepteurs respirent. Les ligands stabilisent des ensembles de conformations. Les endocannabinoid peuvent obtenir une préférence de sous-type grâce à des interactions dynamiques qui modifient la transition de CB1 ou CB2 entre états inactifs, actifs et aptes à la signalisation.
Ce changement de perspective a des conséquences pratiques. Un chimiste médicinal ne demande plus seulement : « Ce composé se lie-t-il à CB1 ou CB2 ? » Les meilleures questions sont : Quelles conformations stabilise-t-il ? Favorise-t-il davantage Gi/o que beta-arrestin ? Déclenche-t-il une internalisation rapide ? Se comporte-t-il différemment dans les neurones, la microglie, les hépatocytes ou les nocicepteurs périphériques ? Peut-il être limité à la périphérie ? Coopère-t-il avec le tonus ligand endogène qui n’augmente qu’en cas d’inflammation ou de lésion ?
La biologie des réseaux pousse cette logique encore plus loin. Une récente analyse intégrative des réseaux, indexée dans PubMed, a identifié CB1 et CB2 comme des nœuds hautement influents au sein du système endocannabinoid et a relié la signalisation des récepteurs à des voies métaboliques plus larges. Cela compte, car la pharmacologie des endocannabinoid ne concerne jamais uniquement les récepteurs. La synthèse, le transport et la dégradation de l’anandamide et du 2-AG modulent l’exposition aux récepteurs. Il en va de même du métabolisme lipidique, des médiateurs inflammatoires et des interactions avec d’autres systèmes GPCR et canaux ioniques. Un ligand qui semble propre dans un test recombinant peut se comporter très différemment dans un tissu vivant où FAAH, MAGL, les voies des prostaglandines et la densité locale des récepteurs varient tous.
Cette vision systémique aide aussi à expliquer pourquoi la traduction clinique a été inégale. Certains échecs sont probablement dus à de mauvais composés. D’autres à des modèles de maladie trop simplistes. Si un essai recrute des patients dont les symptômes partagent une étiquette mais pas une signature endocannabinoid, le résultat moyen peut sembler décevant même si un sous-groupe biologiquement défini pouvait en bénéficier. Les succès futurs dépendront non seulement de la chimie, mais aussi de la stratification : quels patients, quel stade de la maladie, quels tissus, quel état du récepteur, quel tonus endogène.
Des attentes réalistes pour la prochaine génération de thérapeutiques
La prochaine génération de médicaments cannabinoid sera probablement plus étroite, plus sélective et moins romantique que ne le suggère le discours public. C’est une bonne nouvelle. De meilleurs médicaments émergent souvent lorsqu’un domaine renonce aux explications universelles.
Certains progrès sont déjà visibles. La restriction périphérique de CB1 reste attrayante pour la douleur ou les indications métaboliques, car elle vise à réduire les effets indésirables centraux. Les composés ciblant CB2 restent plausibles pour les affections inflammatoires, neuropathiques et neuro-immunes, surtout si l’expression liée à la maladie crée des fenêtres thérapeutiques. Les ligands CB1 biaisés pourraient s’avérer utiles dans les maladies neuropsychiatriques s’ils parviennent à dissocier les effets de circuit souhaités de l’intoxication, de l’anxiété ou de la tolérance. Les modulateurs allostériques peuvent offrir un contrôle plus subtil que les agonistes directs. Les approches ciblant des enzymes qui modifient indirectement le tonus endocannabinoid pourraient encore être utiles, même si elles comportent leurs propres risques et ne contournent pas la complexité des récepteurs.
La prudence reste toutefois essentielle. La sélectivité ne garantit pas le succès. Un agoniste CB2 hautement sélectif peut tout de même échouer si l’expression de CB2 dans la maladie cible est trop variable ou si la signalisation en aval n’est pas le véritable moteur des symptômes. Un biais mesuré dans un essai peut ne pas prédire le biais dans le tissu humain. Les instantanés structuraux sont puissants, mais ce sont des instantanés. La restriction tissulaire peut réduire certaines limites tout en en créant d’autres. Et les phytocannabinoid ne doivent pas être traités comme des substituts de toute la pharmacologie des cannabinoid ; les endocannabinoid, les composés d’origine végétale et les ligands synthétiques peuvent produire des comportements récepteurs très différents selon la dose, le moment et le contexte de signalisation.
Le pronostic sobre est donc le suivant : la future médecine cannabinoid s’améliorera probablement en devenant plus précise, et non en devenant plus globalement activatrice. Les voies les plus crédibles combinent sélectivité des récepteurs, biais de signalisation, contrôle allostérique, ciblage tissulaire et meilleure stratification des patients. Ce que l’on sait est considérable : CB1 et CB2 sont des hubs de signalisation dynamiques, l’état structural compte, et le contexte pathologique compte. Ce qui reste incertain, c’est de savoir si cette clarté mécanistique se traduira en gains cliniques reproductibles dans la douleur, la psychiatrie, l’épilepsie, la neurodégénérescence et les maladies inflammatoires. Le domaine demeure médicalement important parce que le besoin non satisfait est immense, que la biologie est réelle, et que l’ancien modèle simpliste a enfin cédé la place à une pharmacologie capable d’expliquer à la fois la promesse et la déception.
Références
- [1]Cannabis (marijuana). WHO Fact Sheet, 2024. https://www.who.int/news-room/fact-sheets/detail/cannabis-(marijuana)
- [2]Epilepsy. WHO Fact Sheet, 2024. https://www.who.int/news-room/fact-sheets/detail/epilepsy
- [3]Schizophrenia. WHO Fact Sheet, 2022. https://www.who.int/news-room/fact-sheets/detail/schizophrenia
- [4]FDA Regulation of Cannabis and Cannabis-Derived Products, Including Cannabidiol (CBD). FDA Public Health Focus, 2025. https://www.fda.gov/news-events/public-health-focus/fda-regulation-cannabis-and-cannabis-derived-products-including-cannabidiol-cbd
- [5]The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research. National Academies Press, 2017. https://nap.nationalacademies.org/catalog/24625/the-health-effects-of-cannabis-and-cannabinoids-the-current-state