






Why CB1 and CB2 matter in cannabis science
GPCR G protein-coupled receptor: a membrane receptor that changes shape after ligand binding and signals through intracellular partners such as G proteins and beta-arrestins.
What determines receptor output
- Location Tissue, cell type, and subcellular position shape the response.
- Ligand identity Endocannabinoids, phytocannabinoids, and synthetic ligands do not drive identical signaling states.
- Available partners Different cells offer different G proteins, kinases, and beta-arrestins.
- Exposure pattern Signal duration and repeated stimulation affect desensitization and internalization.
- Pathway bias A ligand can favor G-protein signaling, beta-arrestin recruitment, or other outputs.
Public shorthand has long flattened cannabinoid biology into a tidy split: CB1 explains the “high,” CB2 handles inflammation somewhere outside the brain. That framing is too crude to be useful. CB1 and CB2 are G protein-coupled receptors, or GPCRs, and like other GPCRs they do not act as simple on/off switches. They translate signals from endocannabinoids made by the body, phytocannabinoids from Cannabis sativa, and synthetic ligands designed in laboratories into changing cellular responses. Which response appears depends on where the receptor sits, which ligand binds, what signaling partners are available, how long the receptor is stimulated, and whether the receptor is pushed toward G-protein signaling, β-arrestin recruitment, desensitization, or internalization.
That matters because cannabis science is not just about intoxication. It is also about chronic pain, epilepsy, immune signaling, neurodegeneration, psychiatric risk, and why so many cannabinoid drug programs have looked promising preclinically and then stumbled in humans. The stakes are large. The World Health Organization estimated that 200 million people used cannabis in 2019, about 4% of the global population aged 15–64. Epilepsy affects around 50 million people worldwide. Schizophrenia affects about 24 million. And yet, as of 2025, the U.S. FDA notes approval of one cannabis-derived drug product and three cannabis-related drug products. That gap between massive exposure and limited approved therapeutics is one reason receptor biology matters so much.
| Feature | CB1 | CB2 |
|---|---|---|
| Typical shorthand | "brain receptor" | "immune receptor" |
| Article's correction | Central enrichment but also peripheral expression | Immune-enriched but not irrelevant to the brain |
| Example functions mentioned | Perception, memory, motor control, nociception | Cytokine signaling, cell migration, neuroinflammatory roles |
| Interpretation | Circuit- and state-dependent | Cell-type- and disease-state-dependent |
Why receptor biology explains more than plant labels
Labels such as “indica,” “sativa,” or even “THC-dominant” and “CBD-dominant” tell only part of the story because receptors, not plant marketing categories, sit closest to mechanism. Δ9-tetrahydrocannabinol (THC) is a partial agonist at CB1 and CB2, but the downstream effect of THC is not fixed. In cortical neurons rich in CB1, it can suppress neurotransmitter release and alter perception, memory, and motor control. In peripheral sensory pathways, the same receptor family may shape nociception. In immune cells, CB2 activation may shift cytokine signaling or cell migration. Same family. Different outcomes.
The simple rule that CB1 is only brain and CB2 is only immune is too crude for current receptor biology.Strong evidence
The old rule of thumb — CB1 in brain, CB2 in immune cells — came from a real pattern, but it aged badly. Distribution is gradient-based and cell-type-specific, not binary. CB1 is highly expressed in many central nervous system regions, especially on presynaptic terminals, yet it also appears in peripheral tissues. CB2 is strongly associated with immune function, but the claim that it is irrelevant to the brain is no longer defensible. A 2026 review in Frontiers in Behavioral Neuroscience argued that CB2 signaling has gained attention in central nervous system disorders, specifically through neuroinflammatory and neurodegenerative mechanisms, and described this as “an update over the last 3 years.” That update matters. If CB2 contributes to central pathology under some conditions, then drugs aimed at CB2 cannot be understood as purely peripheral tools.
Structure deepens the story. A 2026 Frontiers in Chemical Biology review explained that ligand selectivity between “CB1 and CB2” arises from receptor-level structural differences that influence binding pose, efficacy, and receptor regulation. In plain terms, small chemical changes can bias a ligand toward one receptor subtype or one signaling pathway, which helps explain why two cannabinoids that look similar on paper can feel or perform very differently in vivo. A 2025/2026 PubMed-indexed study on subtype selectivity pushed this further by showing that endocannabinoid selectivity is tied to receptor conformational dynamics rather than a rigid lock-and-key model. The receptor moves. The ligand stabilizes some states more than others. Biology follows those states.
From phytocannabinoids to endocannabinoid signaling
{kind=link}
Retrograde signaling A synaptic signaling pattern in which a postsynaptic cell releases a messenger that travels backward to act on presynaptic receptors.
Discovery sequence in brief
- 1 THC was isolated and structurally defined.
- 2 Specific cannabinoid binding sites were demonstrated in brain tissue.
- 3 CB1 was cloned as a GPCR.
- 4 CB2 was identified from immune-related tissues.
- 5 Anandamide and then 2-AG established an endogenous signaling system.
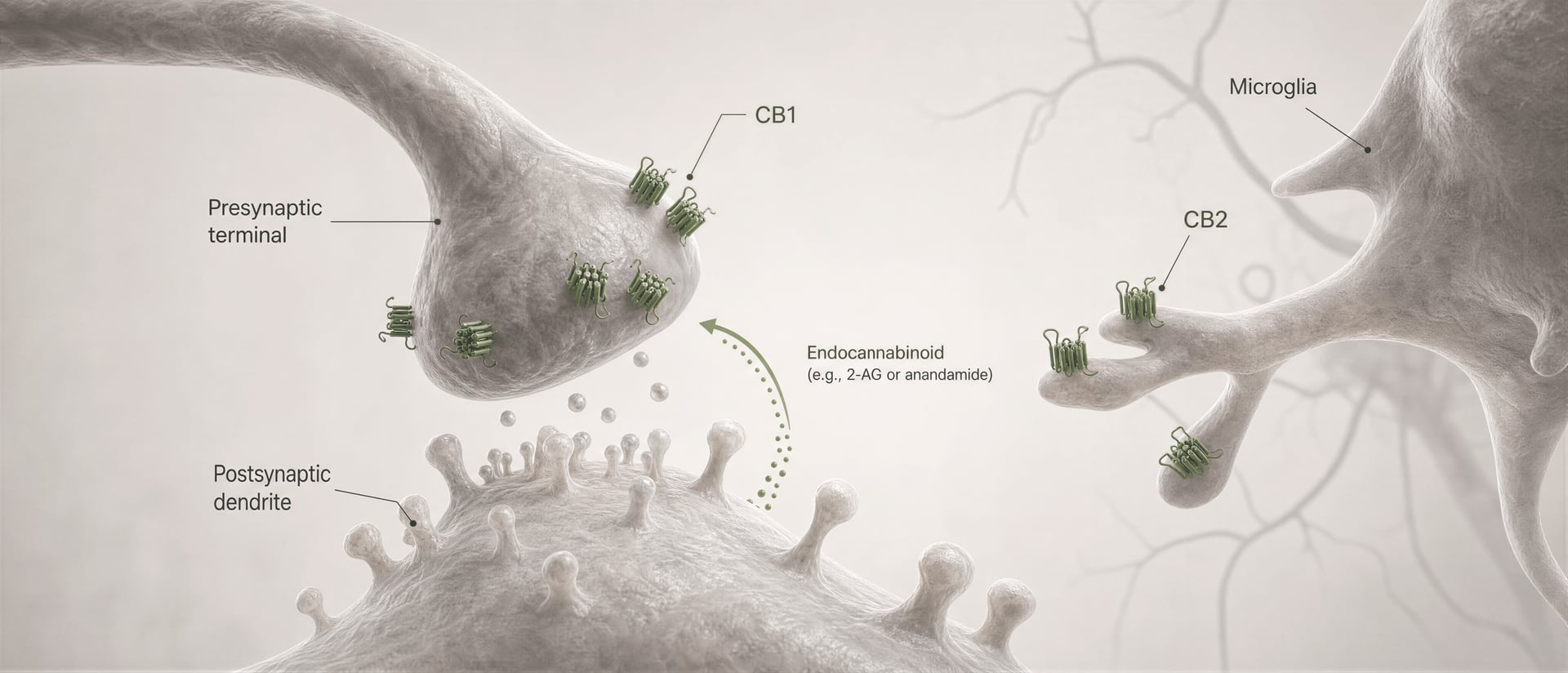
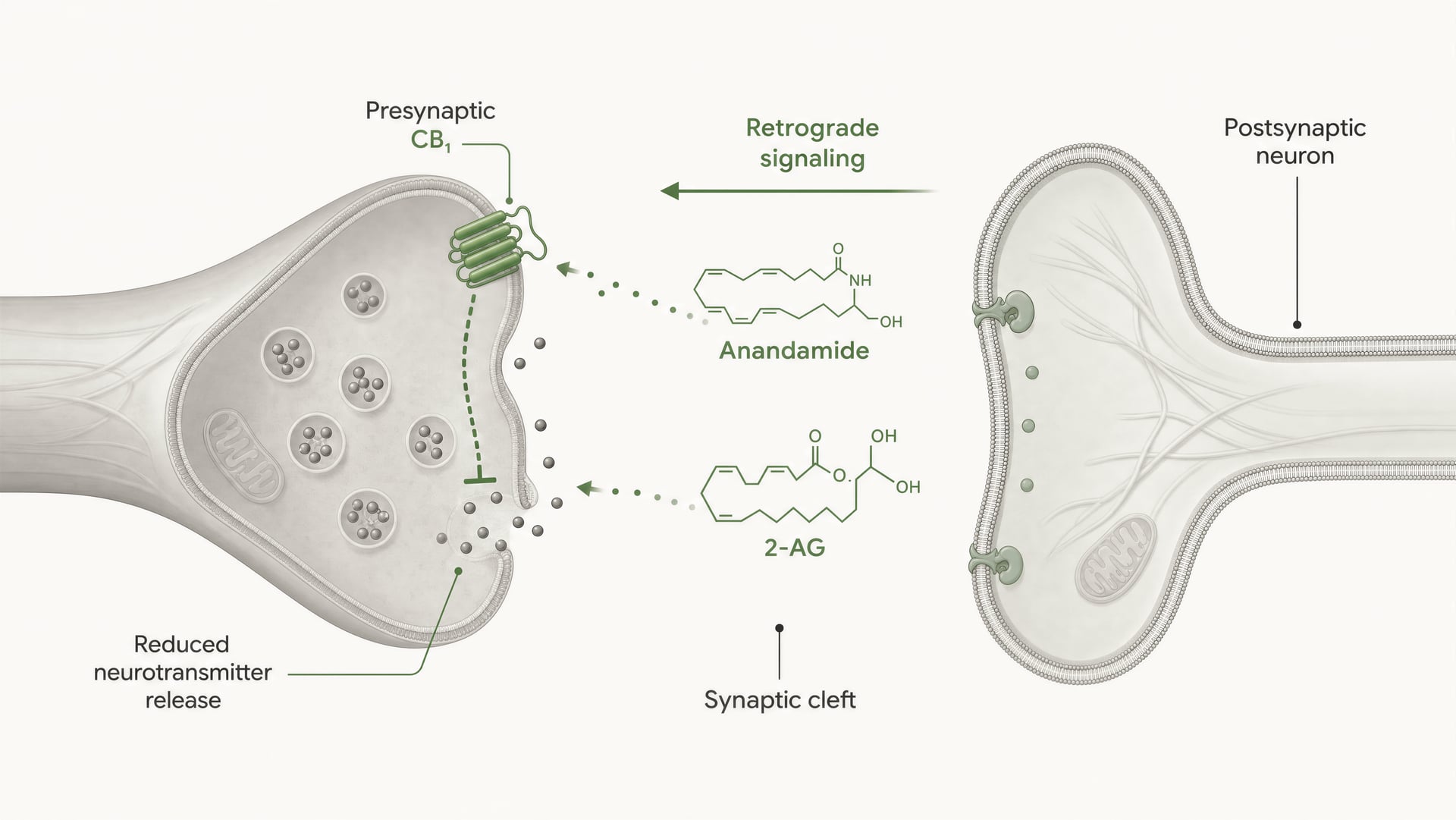
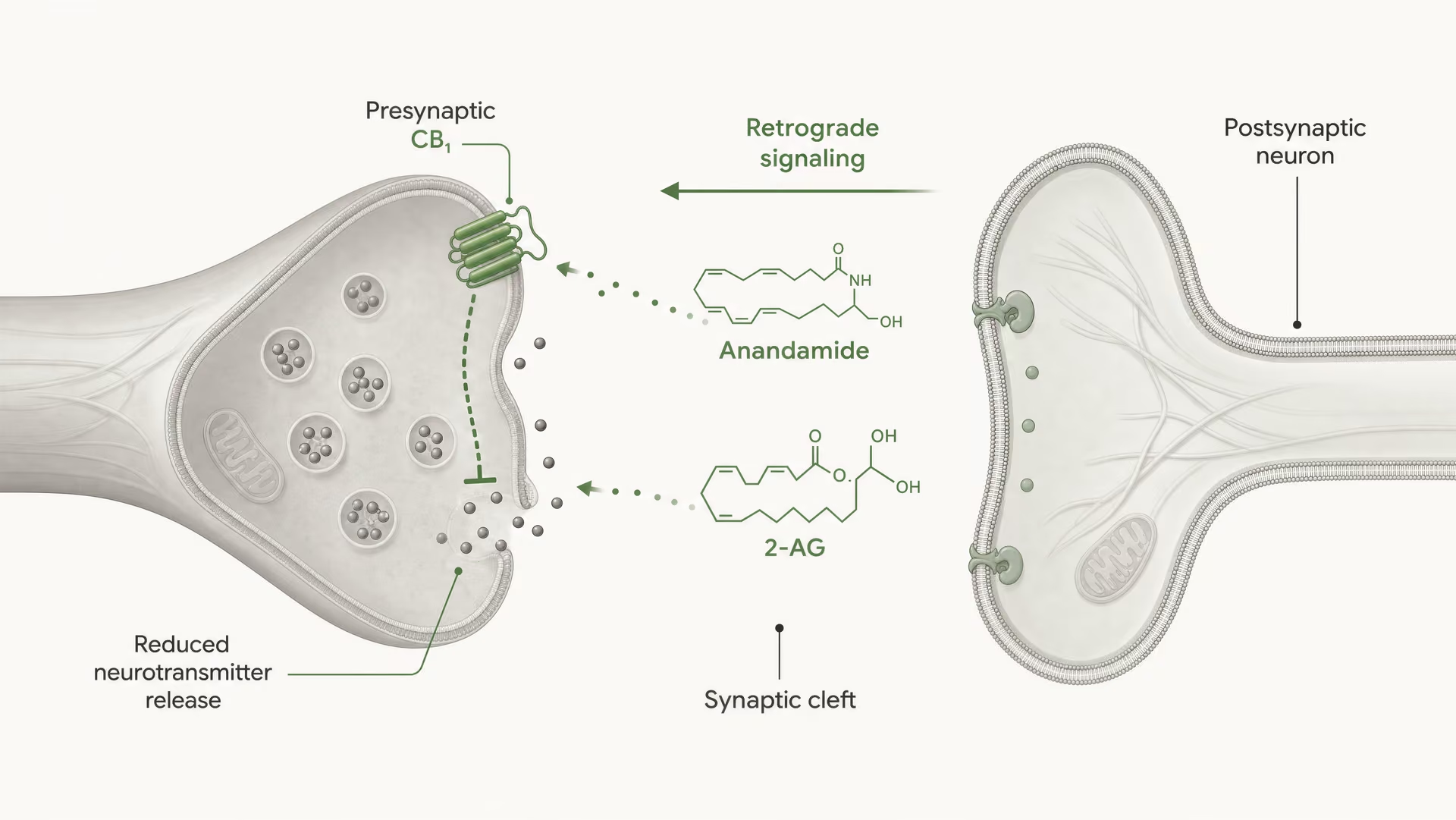
Cannabinoid receptors were not discovered because the body evolved for cannabis. The sequence ran the other way. Work by Allyn Howlett and colleagues was central to defining cannabinoid receptor pharmacology, and the later discovery of anandamide by Raphael Mechoulam and Lumír Hanuš helped establish that humans make their own cannabinoid-like signaling molecules. Anandamide and 2-arachidonoylglycerol, usually shortened to 2-AG, are the main endocannabinoids. They are produced on demand, not stored in vesicles like classical neurotransmitters, and often signal retrogradely: a postsynaptic cell generates an endocannabinoid that travels backward across the synapse to activate presynaptic CB1 and reduce further transmitter release.
That is a fundamentally different picture from “weed chemical hits receptor.” Endocannabinoid signaling is local, transient, and tightly regulated by synthesis and degradation enzymes. Phytocannabinoids enter that system from outside. Synthetic ligands can hit it even harder or more selectively. The result is that the same receptor may be engaged by a fleeting endogenous pulse, a slowly absorbed oral phytocannabinoid, or a high-efficacy synthetic agonist with very different safety liabilities.
This difference is one reason intoxication cannot be inferred from receptor name alone. It depends on ligand efficacy, dose, route, timing, and tissue context. THC at CB1 is central to psychoactive effects, yes, but that fact does not reduce CB1 to a “psychoactivity receptor.” Nor does it make CB2 a simple anti-inflammatory dial. The 2025 American Journal of Psychiatry article on CB1 biased signaling made exactly this broader point by arguing that CB1-biased ligands may offer a therapeutic strategy for schizophrenia. That proposal links cannabinoid science to a wider GPCR idea: if one ligand favors beneficial signaling branches while avoiding others tied to adverse effects, drug action may be separable from blunt receptor activation. Whether that promise holds clinically remains unsettled, but the mechanistic argument is strong.
What this article means by distribution, signaling, and drug targets
In this article, distribution means more than an organ map. It includes receptor density, cell type, subcellular location, disease state, and temporal change. A receptor expressed on GABAergic terminals can have different circuit effects from the same receptor on glutamatergic terminals. A receptor upregulated during inflammation is not equivalent to its baseline state. Distribution is dynamic.
Signaling means the intracellular consequences of receptor engagement. For CB1 and CB2, that includes coupling to Gi/o-family G proteins, inhibition of adenylyl cyclase, modulation of ion channels, changes in kinase cascades, β-arrestin recruitment, receptor desensitization, and internalization. It also includes allosteric modulation and biased agonism, where ligands can favor some signaling outputs over others. This is not academic fine print. It is often the difference between analgesia, sedation, tolerance, dysphoria, or a failed trial.
Drug targets means receptors considered for intervention, not guaranteed success stories. Selective CB1 targeting might reduce some off-target effects yet still run into central adverse events. Selective CB2 targeting may avoid some intoxicating liabilities, but selectivity does not guarantee efficacy in complex human disease. Systems biology work makes that clear. A 2025/2026 PubMed-indexed integrative network analysis identified CB1 and CB2 as highly influential nodes in the endocannabinoid system and linked their signaling to broader metabolic pathways. In other words, these receptors sit inside larger networks. Push one node, and other pathways move.
That is the stance of this article. CB1 and CB2 are context-dependent signaling nodes. Not static switches. Not mere labels for “brain” and “immune system.” If cannabis science is going to explain why a compound looks therapeutic in one setting, intoxicating in another, and disappointing in a clinic, it has to start at the receptor level and stay there long enough to follow the biology where it actually goes.
A brief history of cannabinoid receptor discovery
Before cannabinoid receptors were identified, cannabis science was mostly a chemistry story. Researchers could isolate plant compounds, compare crude behavioral effects in animals, and argue about potency, but they could not yet explain how a molecule such as delta-9-tetrahydrocannabinol, or THC, produced its effects with anything like receptor-level precision. That changed in the late 1980s and early 1990s. The shift was decisive: cannabis research moved from cataloging phytocannabinoids to studying ligand-receptor interactions, intracellular signaling, tissue distribution, and eventually the endogenous lipid system now called the endocannabinoid system, or ECS.
| Year | Milestone | People named in article |
|---|---|---|
| 1964 | Isolation and structure of THC | Raphael Mechoulam; Yechiel Gaoni |
| 1988 | Specific high-affinity cannabinoid binding sites in rat brain membranes | Allyn Howlett; William Devane |
| 1990 | Cloning of CB1 | Lisa Matsuda and colleagues |
| 1992 | Identification of anandamide | William Devane; Lumír Hanuš; Raphael Mechoulam; colleagues |
| 1993 | Identification of CB2 | Sean Munro; Kerrie Thomas; M. Abu-Shaar |
| 1995 | Identification of 2-AG by separate groups | Mechoulam team; Sugiura group |
From THC pharmacology to receptor identification
A key early milestone came in 1964, when Raphael Mechoulam and Yechiel Gaoni reported the isolation and structure of THC. That achievement mattered because it gave pharmacologists a defined molecule to test rather than a variable botanical extract. For the next two decades, the field built a structure-activity map from THC and related cannabinoids, but there was still debate over mechanism. Some investigators favored nonspecific membrane effects because cannabinoids are lipophilic. That view became harder to defend as stereoselective and saturable binding data accumulated.
The receptor era properly began with binding studies in the 1980s. In 1988, Allyn Howlett and William Devane published a landmark paper in Molecular Pharmacology showing specific, high-affinity cannabinoid binding sites in rat brain membranes using the synthetic agonist CP55,940. The result was not a vague suggestion of a target. It showed saturability, regional variation, and pharmacological specificity consistent with a bona fide receptor. Brain tissue was not responding to cannabinoids as if they were simply dissolving into lipid bilayers and perturbing everything at once. There was selectivity.
Three years later, in 1990, Lisa Matsuda and colleagues cloned the first cannabinoid receptor, now called CB1, and published it in Nature. CB1 was identified as a G protein-coupled receptor, or GPCR, a finding that immediately placed cannabinoid pharmacology inside one of the most important signaling superfamilies in biology. That mattered because GPCRs are not just switches. They adopt multiple conformational states, couple to different intracellular partners, desensitize, internalize, and show ligand-dependent signaling bias. Those ideas would become central much later, but the cloning of CB1 made them possible.
CB2 followed quickly. In 1993, Sean Munro, Kerrie Thomas, and M. Abu-Shaar identified a second cannabinoid receptor, CB2, also in Nature, first characterized from immune-related tissues. This discovery created a durable shorthand that shaped the field for years: CB1 as the “brain receptor” associated with intoxication, CB2 as the “peripheral” or immune receptor associated with inflammation. The shorthand was useful, but it was also too crude even then, and it has aged badly. The distribution of both receptors depends on species, cell type, activation state, disease context, and assay method.
How CB1 and CB2 changed the endocannabinoid field
Once CB1 and CB2 were identified, the obvious next question was why the body had receptors for plant-derived cannabinoids at all. The answer arrived in 1992, when William Devane, Lumír Hanuš, Raphael Mechoulam, and colleagues identified anandamide, formally arachidonoyl ethanolamide, as an endogenous ligand. The paper, published in Science, marked a conceptual break. Cannabis pharmacology was no longer just about exogenous compounds from Cannabis sativa. It was about a native lipid signaling system.
A second major endogenous ligand, 2-arachidonoylglycerol or 2-AG, was identified in 1995 by separate groups including Mechoulam’s team and Sugiura’s group. With receptors and endogenous ligands in place, the ECS expanded rapidly. Researchers identified synthetic and degradative enzymes such as fatty acid amide hydrolase, FAAH, for anandamide and monoacylglycerol lipase, MAGL, for 2-AG. They also confronted a still unsettled issue: how these highly lipophilic molecules move across membranes and extracellular space. The field often speaks of “transport,” but a single dedicated endocannabinoid transporter has remained elusive.
This was the point at which cannabinoid science stopped being a two-receptor chart and became a signaling network. CB1 and CB2 were linked to Gi/o proteins, inhibition of adenylyl cyclase, modulation of calcium and potassium channels, and suppression of transmitter release. But the story did not stay that simple. Receptors could recruit beta-arrestins, undergo desensitization and internalization, and respond differently to phytocannabinoids, endocannabinoids, and synthetic ligands even when those ligands nominally hit the same receptor. The current GPCR language of biased agonism fits cannabinoids especially well. A 2025 American Journal of Psychiatry article argued that CB1 biased signaling is a plausible therapeutic strategy for schizophrenia, a disorder that affects about 24 million people worldwide according to the WHO. That is a long way from the old image of CB1 as merely the receptor that explains why THC intoxicates.
The CB2 story has also widened. Early work placed it mainly in immune tissues, and that was directionally correct, but later studies found CB2 expression in microglia and under some conditions in other central nervous system cell populations. A 2026 Frontiers in Behavioral Neuroscience review described “an update over the last 3 years” linking CB2 signaling to neuroinflammatory and neurodegenerative mechanisms, making clear that CB2 cannot be dismissed as irrelevant to the brain. Current structural work has gone further still. A 2026 Frontiers in Chemical Biology review on “CB1 and CB2” emphasized that subtype selectivity depends on receptor-level structural differences that alter binding, efficacy, and regulation. A recent PubMed-indexed study on subtype selectivity likewise argues that endocannabinoid selectivity is dynamic, shaped by conformational behavior rather than a simple lock-and-key model.
Foundational researchers and why the history still matters
Three names belong near the center of this history. Raphael Mechoulam helped define the chemical and biological basis of cannabinoid science, from THC structure work to endocannabinoid discovery. Lumír Hanuš was a central figure in the identification of anandamide and later endocannabinoid research. Allyn Howlett’s receptor pharmacology was decisive in proving that cannabinoids act through specific brain binding sites and signaling mechanisms. Without their work, there is no modern ECS field.
The history still matters because old simplifications continue to distort present debates. In 2019, an estimated 200 million people worldwide, about 4% of those aged 15 to 64, used cannabis according to the WHO. At the same time, the FDA states that it has approved one cannabis-derived drug product and three cannabis-related drug products. Public exposure is huge. Clinical translation is selective and difficult. Receptor history explains why. Cannabinoid effects depend on ligand class, receptor state, tissue localization, timing, and pathway bias. They also depend on a broader network. A 2025/2026 integrative network analysis identified CB1 and CB2 as highly influential nodes connected to metabolic pathways rather than isolated targets.
That is the real legacy of receptor discovery. It did not simplify cannabis biology. It showed why the biology is harder than the old brain-versus-body split ever allowed.
Where CB1 is found: brain circuits, peripheral tissues, and functional gradients
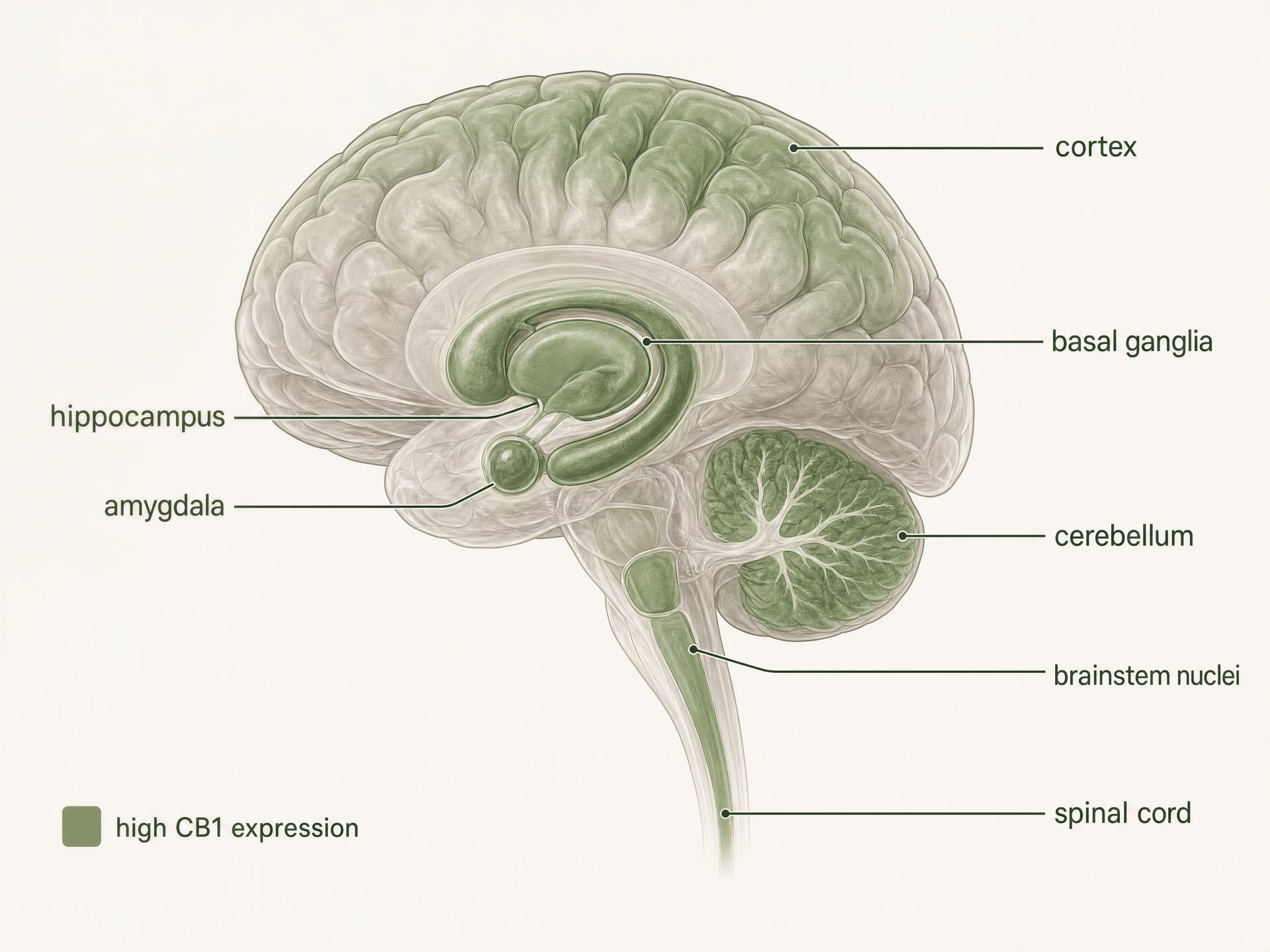
CB1 earned its reputation as the main psychoactive cannabinoid receptor for a reason. It is abundant in the central nervous system, and Allyn Howlett’s receptor pharmacology work helped establish that THC acts through a specific, saturable receptor system rather than through nonspecific membrane effects. But the old shorthand — CB1 in brain, CB2 in immune cells — now causes more confusion than clarity. CB1 is strongly enriched in neural circuits, yes. It is also present in the gut, liver, adipose tissue, reproductive organs, cardiovascular tissues, and sensory pathways, where it shapes feeding, metabolism, pain signaling, and autonomic function. Distribution is broad. Function is conditional.
That matters because cannabinoid exposure is common. The World Health Organization estimated that 200 million people used cannabis in 2019, about 4% of the global population aged 15–64. It also matters because receptor pharmacology keeps spilling into medicine: the U.S. FDA states that one cannabis-derived drug product and three cannabis-related drug products are approved. A receptor found across so many organs cannot be reduced to a single behavioral label.
- General pattern
- One of the most abundant GPCRs in the mammalian brain
- High-density regions named
- Cortex, hippocampus, amygdala, basal ganglia, cerebellum
- Pain-related sites named
- Periaqueductal gray, rostral ventromedial medulla, dorsal horn
- Peripheral sites named
- Gut, liver, adipose tissue, reproductive, cardiovascular, sensory pathways
High expression in the central nervous system
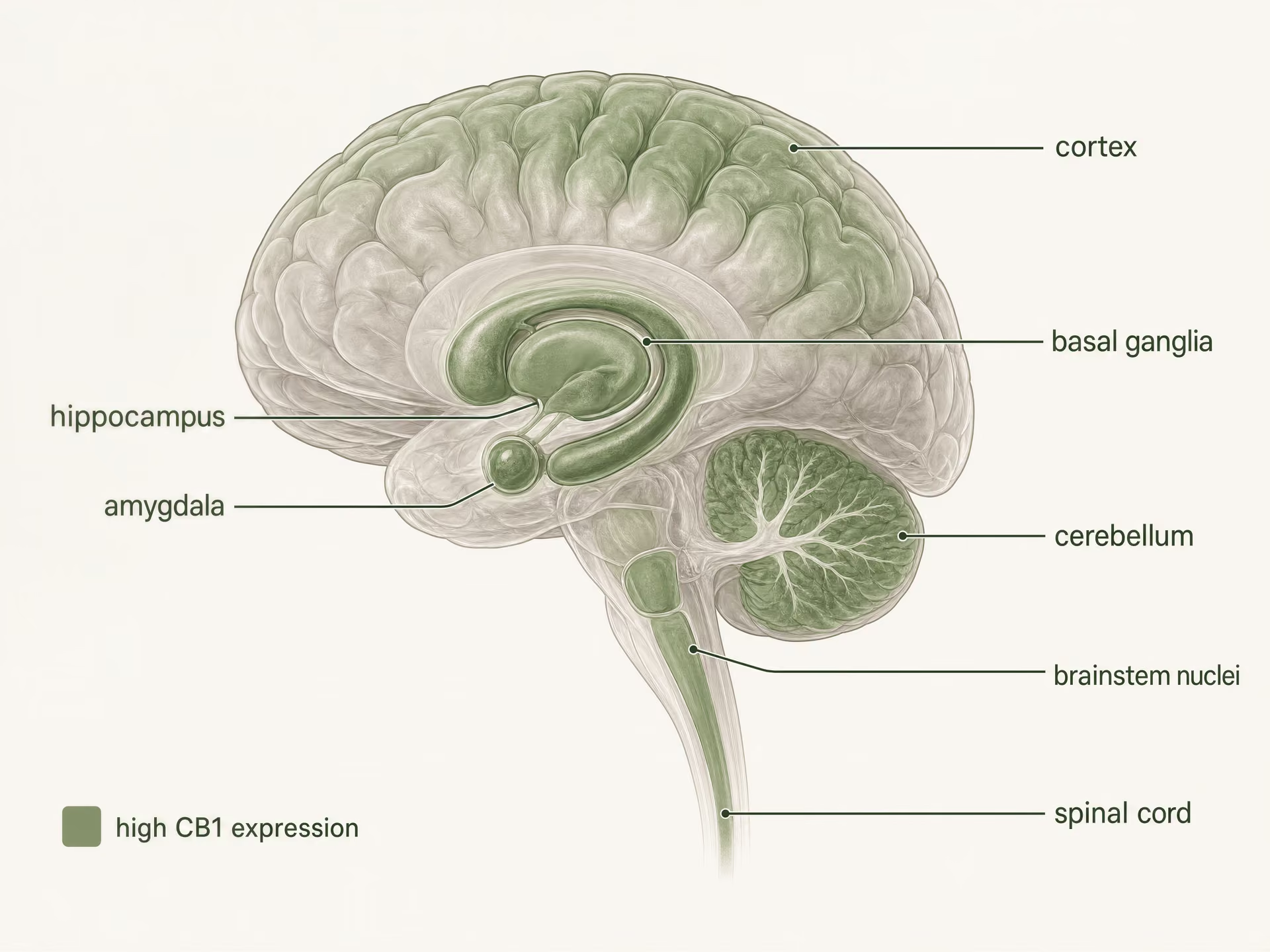{kind=link}
CB1 is one of the most abundant G protein-coupled receptors in the mammalian brain. Autoradiography, in situ hybridization, and immunohistochemical mapping built a clear picture long before current structural studies: high densities appear in cortex, hippocampus, amygdala, basal ganglia, cerebellum, and several pain-processing regions, with additional expression in brainstem nuclei and throughout the spinal cord. This pattern fits the classic effects of THC surprisingly well, but not perfectly.
| CB1 location | Immediate synaptic effect | Example consequence named in article |
|---|---|---|
| GABAergic interneuron terminals | Suppresses GABA release | Disinhibition of downstream neurons |
| Glutamatergic terminals | Suppresses glutamate release | Dampened excitation |
| Basal ganglia and cerebellar circuits | Alters transmitter release in motor pathways | Motor slowing, altered habit circuitry, impaired coordination |
| Pain pathways | Modulates nociceptive transmission | Changes in ascending, descending, inflammatory, and affective pain processing |
In the cortex and hippocampus, CB1 sits in circuits that regulate attention, working memory, extinction learning, and short-term synaptic plasticity. Memory effects are not just “hippocampus equals forgetfulness.” They depend heavily on which axon terminals express the receptor. CB1 is often concentrated presynaptically, where it suppresses neurotransmitter release after activation by endocannabinoids such as anandamide and 2-arachidonoylglycerol, the signaling lipids whose discovery by Raphael Mechoulam, Lumír Hanuš, and colleagues transformed the field. When CB1 is engaged on GABAergic interneuron terminals, it can disinhibit downstream neurons; when engaged on glutamatergic terminals, it can dampen excitation. Same receptor, opposite network result.
The basal ganglia and cerebellum explain another familiar set of effects. Dense CB1 expression in striatum, globus pallidus, substantia nigra pars reticulata, and cerebellar molecular layers links receptor activation to motor slowing, altered habit circuitry, impaired coordination, and, at some doses, catalepsy-like effects in animal models. Yet the fact that CB1 is sparse in the brainstem cardiorespiratory centers compared with opioid receptors helps explain why cannabinoid overdose does not typically produce the same fatal respiratory depression pattern seen with strong opioid agonists. Location matters. So does what is absent.[5]The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research. National Academies of Sciences, Engineering, and Medicine. National Academies Press, 2017. https://nap.nationalacademies.org/catalog/24625/the-health-effects-of-cannabis-and-cannabinoids-the-current-state
Pain processing shows the same regional logic. CB1 is found in the periaqueductal gray, rostral ventromedial medulla, dorsal horn of the spinal cord, and peripheral nociceptive pathways. That gives the receptor multiple entry points into nociception: it can alter ascending pain signals, descending pain control, inflammatory sensitization, and the emotional coloring of pain. This is one reason cannabinoids have remained in the chronic pain discussion, especially since nearly 1 in 5 adults in the United States live with chronic pain, according to the National Academies. But analgesia is not guaranteed simply because CB1 is present. Sedation, cognitive impairment, tolerance, and dose-limiting adverse effects often arrive through nearby circuits or through the same circuits at different levels of receptor engagement.
CB1 biased signaling may separate desired therapeutic effects from unwanted psychoactive or cognitive effects.Limited evidence
Gi/o proteins A family of G proteins that commonly reduce adenylyl cyclase activity and help control ion channels after GPCR activation.
Modern receptor biology adds another layer. CB1 is not a simple on-off switch. It primarily couples to Gi/o proteins, reducing adenylyl cyclase activity and modulating ion channels, but it can also recruit beta-arrestins, undergo desensitization and internalization, and display ligand-dependent signaling bias. The 2025 American Journal of Psychiatry article arguing that CB1 biased signaling could be exploited therapeutically in schizophrenia makes this point directly: receptor occupancy alone is a poor predictor of outcome. With schizophrenia affecting about 24 million people worldwide, the appeal of separating desired signaling from unwanted psychoactive or cognitive effects is obvious. Whether that separation is achievable in practice remains an open drug-development question, not a settled fact.
Peripheral CB1 in gut, liver, adipose tissue, and beyond
CB1 outside the brain is not a footnote. It is central to why cannabinoids affect appetite, nausea, glucose handling, lipid metabolism, and visceral sensation.
In the gut, CB1 is expressed in the enteric nervous system, epithelial compartments, and vagal-related pathways. Activation can slow gastric emptying, alter intestinal motility, reduce emesis, and change signaling between the gut and brain. Appetite effects are often described as if they originate purely in hypothalamic reward and feeding centers, but peripheral CB1 contributes to the story by shaping sensory and hormonal inputs before signals even reach those circuits. A meal does not act on a blank receptor landscape; it changes endocannabinoid tone locally.
In liver and adipose tissue, CB1 participates in metabolic regulation, including lipogenesis, insulin sensitivity, and energy storage. This was one of the major lessons of the rimonabant era. Blocking CB1 improved weight and metabolic markers, which supported the idea that overactive endocannabinoid signaling contributes to obesity-related pathology. But rimonabant, a centrally active CB1 inverse agonist, also produced serious psychiatric adverse effects, including depression and anxiety, and was withdrawn. That episode is often cited as a failure of “CB1 targeting.” More accurately, it was a failure of a particular kind of CB1 targeting: strong central antagonism or inverse agonism in a receptor system embedded in mood and stress circuits. The lesson is not that peripheral CB1 is irrelevant; it is that drug exposure pattern and receptor state matter as much as receptor name.
Adipocytes, hepatocytes, pancreatic tissue, skeletal muscle, cardiovascular tissues, and reproductive organs all add to the peripheral map. So do sensory neurons. The 2025/2026 PubMed-indexed integrative network analysis that identified CB1 and CB2 as highly influential nodes in endocannabinoid signaling is useful here because it shifts the frame from receptor location alone to receptor participation in metabolic and signaling networks. A receptor with modest expression in one tissue can still exert large system-level effects if it sits at a bottleneck in local signaling.
Structural work also keeps this discussion honest. The 2026 Frontiers in Chemical Biology review on CB1 and CB2 emphasizes that ligand selectivity and efficacy arise from receptor-level structural differences that alter binding, signaling, and receptor regulation. A 2025/2026 PubMed-indexed study on subtype selectivity likewise argues that conformational dynamics, not just lock-and-key fit, shape how endocannabinoids distinguish receptor subtypes. That matters for CB1 distribution because “CB1 in liver” does not mean THC, anandamide, 2-AG, and a synthetic agonist will all do the same thing there.
Why distribution does not equal a single uniform function
The biggest mistake in receptor maps is to treat expression as destiny. It is not. High expression tells you where to look, not what will happen.
First, cell type changes the sign of the effect. A CB1 receptor on a glutamatergic terminal can reduce excitation. The same receptor on a GABAergic terminal can reduce inhibition. Those are not interchangeable outcomes. Second, synaptic location matters. CB1 is usually presynaptic, often engaged by endocannabinoids released “on demand” from postsynaptic neurons, creating retrograde control over neurotransmitter release. That arrangement favors brief, activity-dependent modulation rather than constant receptor activation.
Third, ligand identity matters. Endocannabinoids are short-lived local messengers. Phytocannabinoids such as THC arrive from outside the system, often at higher and longer-lasting exposures than endogenous signals. Synthetic ligands can push even harder, with different efficacy and bias. Some promote Gi/o signaling more strongly; others favor beta-arrestin recruitment, desensitization, or receptor internalization. That is why two compounds can both be called CB1 agonists yet differ sharply in appetite stimulation, memory disruption, motor impairment, and tolerance development.
Fourth, local ligand availability changes everything. Anandamide and 2-AG are made and degraded on site, so their effects depend on neural activity, metabolic state, enzyme expression, and inflammatory context. Fifth, receptor density itself exists on a gradient. Brain region, developmental stage, disease state, and repeated drug exposure all shift CB1 levels and responsiveness.
The current literature is moving away from binaries for exactly this reason. The 2026 Frontiers in Behavioral Neuroscience review notes an update over the last 3 years in how cannabinoid receptor signaling is understood in CNS disorders, especially once neuroinflammatory and neurodegenerative mechanisms are included. CB1 should be read with the same caution. It is a dominant central receptor, but not an exclusively central one; a feeding receptor, but not only that; a pain target, but not a clean analgesic switch. Any serious account of CB1 distribution has to think in gradients, circuits, and signaling states rather than in a brain-versus-body cartoon.
Where CB2 is found: immune system roots and the expanding CNS map
The old shorthand said it cleanly: CB1 is the brain receptor, CB2 is the immune receptor. That framing helped early teaching, but it now misleads more than it clarifies. CB2 does show classical enrichment outside neurons, especially across immune and hematopoietic lineages, and that fact still matters for pharmacology. Yet the newer literature, especially the 2026 Frontiers in Behavioral Neuroscience review, makes a stronger claim: CB2 is now being discussed in central nervous system disorders because its expression and signaling become more visible in microglia, inflammatory circuits, and injury-related states, an “update over the last 3 years” that has changed how the receptor is mapped and interpreted. The result is not that CB2 suddenly became a high-abundance, pan-neuronal brain receptor. It did not. The result is that the receptor’s distribution has to be described as conditional, cell-specific, and state-dependent.
That distinction matters clinically. The World Health Organization estimated that 200 million people used cannabis in 2019, or 4% of the global population aged 15–64. Even with only a small number of approved cannabinoid-related medicines—the FDA in 2025 counted one cannabis-derived product and three cannabis-related products—receptor localization still shapes where drug developers look for anti-inflammatory, analgesic, neuroprotective, and psychiatric effects, and where they expect liabilities.
- Classical enrichment
- Immune and hematopoietic cells
- Cell types named
- B cells, T cells, macrophages, monocytes, natural killer cells, neutrophils, mast cells
- Canonical tissues named
- Spleen, tonsil, thymus, bone marrow, circulating immune cells
- CNS relevance emphasized
- Microglia and pathology-linked states
Classical enrichment in immune and hematopoietic cells
CB2 was originally identified as the cannabinoid receptor subtype with strongest expression in cells tied to immunity rather than fast synaptic transmission. That remains the right starting point. Compared with CB1, which is heavily represented across many neuronal populations, CB2 is classically enriched in B cells, T cells, macrophages, monocytes, natural killer cells, neutrophils, mast cells, and other hematopoietic compartments. Spleen, tonsil, thymus, bone marrow, and circulating immune-cell populations have therefore been the canonical tissues for CB2 analysis.
This immune weighting shaped the early drug-development idea of CB2-selective agonists as a way to capture anti-inflammatory or analgesic benefits while avoiding the intoxicating effects associated with strong CB1 activation in brain. It was a sensible hypothesis, but only half-complete. CB2 is a Gi/o-coupled GPCR, and like CB1 it does not merely switch “on” or “off.” Depending on ligand, receptor conformation, and cellular context, CB2 can reduce adenylyl cyclase activity, influence MAPK pathways, alter ion-channel coupling indirectly, recruit beta-arrestins, and undergo desensitization or internalization. So even in peripheral immune tissues, the real question is not just whether CB2 is present, but which cells express it, at what level, under what stimulus, and with what downstream bias.
That complexity is one reason similar-looking ligands can behave differently. The 2026 Frontiers in Chemical Biology review on cannabinoid receptor structure argues that selectivity at “CB1 and CB2” is shaped by receptor-level structural differences that alter ligand binding, signaling efficacy, and receptor regulation. A 2025/2026 PubMed-indexed study on subtype selectivity pushed the point further by showing that endocannabinoid selectivity is dynamic and tied to conformational states, not a rigid lock-and-key model. That matters for tissue mapping because an endogenous ligand such as 2-AG or anandamide, a phytocannabinoid such as THC, and a synthetic CB2-preferring agonist may all encounter the same receptor population but stabilize different signaling outputs.
The older immune-centered map of CB2 was therefore not wrong. It was incomplete. CB2 is still best described as a receptor with strong immune-system roots. But roots are not the whole organism.
| Context | How CB2 is described | Interpretive point |
|---|---|---|
| Healthy brain baseline | Often low or near detection limits in many regions | Low basal signal does not equal no relevance |
| Activated microglia | More detectable after injury or inflammation | Supports CNS relevance through immune-like functions |
| Astrocytes / endothelial / infiltrating cells | Reported in some disease contexts | Localization depends on method and model |
| Broad constitutive neuronal expression | Needs stronger evidence | The article treats this claim cautiously |
CB2 in microglia, neuroinflammation, and injury states
The strongest case for central relevance does not come from claiming that CB2 is broadly abundant on healthy forebrain neurons. It comes from microglia and from disease biology.
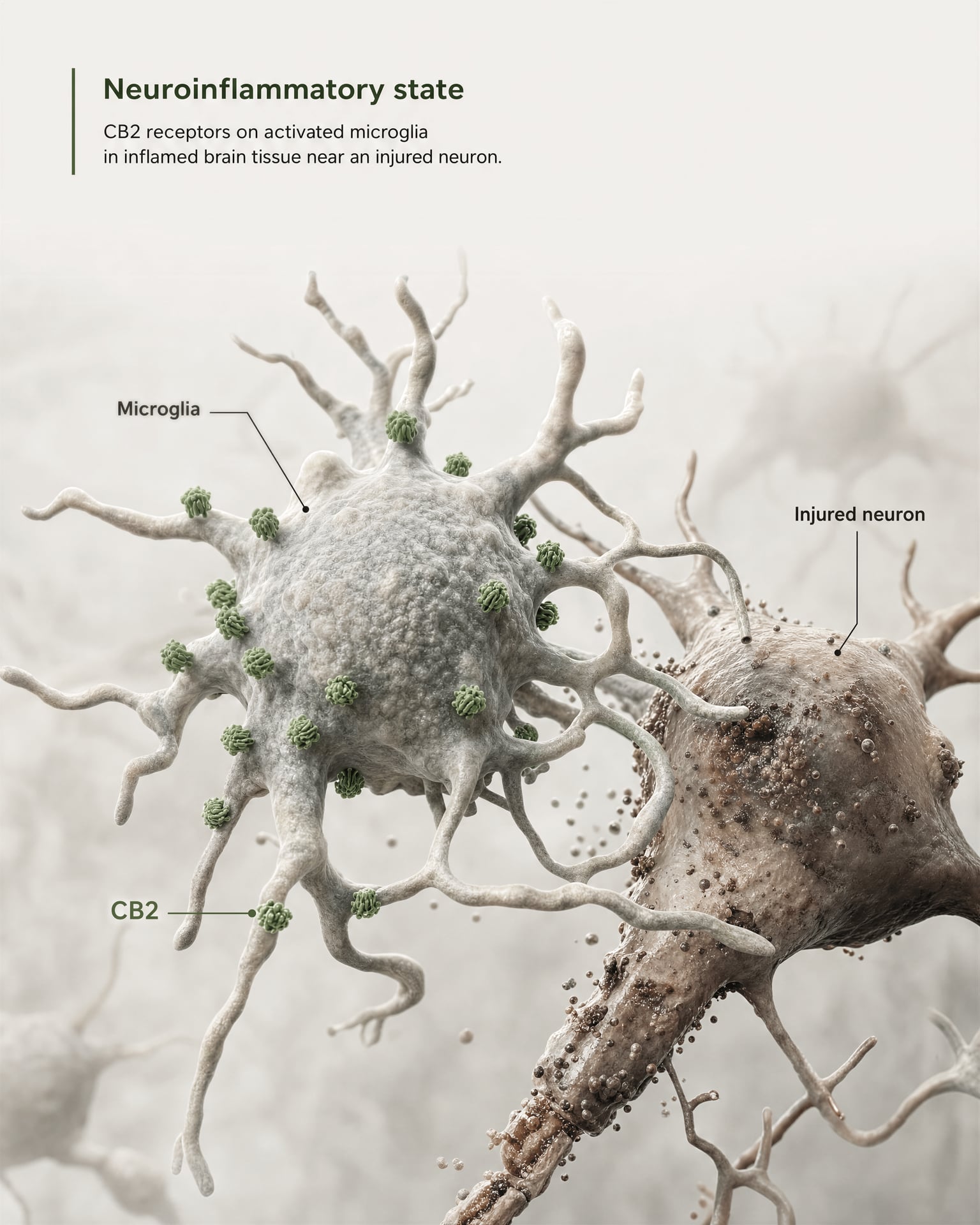
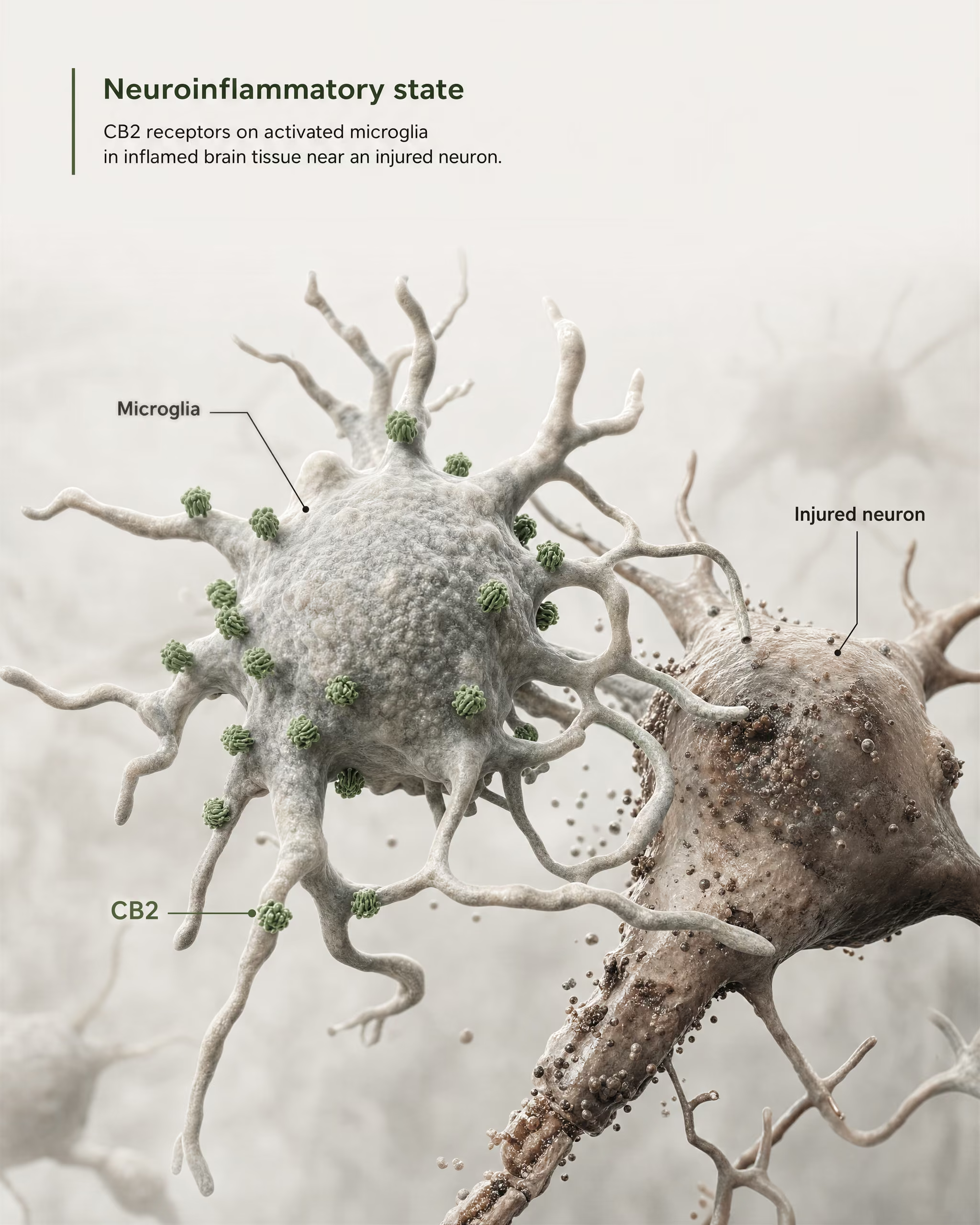{kind=link}
Microglia are the resident immune cells of the CNS, and they sit exactly at the boundary where the old “peripheral immune receptor” model starts to fail. If a receptor is expressed in the brain’s own immune surveillance and inflammatory response system, then calling it merely peripheral becomes inaccurate. The 2026 Frontiers in Behavioral Neuroscience review makes this point directly: CB2 signaling has gained attention in CNS disorders because it links to neuroinflammatory and neurodegenerative mechanisms. That is why CB2 now appears in discussions of Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, traumatic brain injury, stroke, neuropathic pain, and some psychiatric conditions where inflammatory signaling is part of the pathology.
The key phrase is induced or increased expression. In many healthy brain regions, basal CB2 expression is low, sometimes near the limits of older detection methods. But after injury, infection, chronic inflammation, or neurodegeneration, CB2 signal often becomes more detectable, particularly in activated microglia and, in some studies, astrocytes, infiltrating immune cells, endothelial compartments, or restricted neuronal subsets. This is a very different distribution rule than the one usually applied to CB1. CB1 is often constitutively abundant in defined neuronal circuits. CB2 is more often interpreted as a receptor whose CNS relevance emerges under stress, pathology, or inflammatory activation.
That distinction has practical consequences. A CB2-targeted drug may have little effect in a healthy tissue where receptor density is low, yet show measurable activity in a diseased tissue where expression has risen and signaling networks have changed. This inducibility is one reason preclinical findings have been both exciting and hard to translate. Timing matters. Disease stage matters. Cell composition matters. A post-injury microglial environment is not pharmacologically equivalent to an unstimulated brain slice.
The interpretive problems are not trivial. CB2 has a long history of antibody specificity concerns, low-abundance transcript detection, species differences, and inconsistent localization claims across methods. Some early reports likely overstated neuronal CB2 because available tools were weak. That is why careful studies now lean on converging evidence—single-cell transcriptomics, in situ hybridization, validated genetic reporters, knockout controls, proteomic data where possible, and state-dependent comparisons—rather than a single staining result. If one study reports CB2 in neurons at baseline and another fails to detect it, the discrepancy may reflect true regional differences, disease status, species, age, or simply assay limitations.
CB2 has meaningful CNS relevance, especially in glial and injury-linked inflammatory contexts.Limited evidence
So the best current position is restrained but clear: CB2 has real CNS relevance, mainly through glial and immune-like functions, and this relevance increases during neuroinflammation and injury. Claims of broad constitutive neuronal CB2 expression in the normal brain need stronger evidence than claims of microglial and pathology-associated CB2.
How the last 3 years changed the CB2 conversation
The 2026 Frontiers in Behavioral Neuroscience review explicitly frames the recent literature as an “update over the last 3 years,” and that wording captures a real shift. The conversation moved away from arguing over whether CB2 is “in the brain at all” and toward asking where, when, and in which disease states its signaling becomes actionable.
Three developments drove that shift. First, cell-resolution methods improved. Single-cell and single-nucleus RNA datasets, better spatial mapping, and stricter validation standards reduced the chance that low-level or inducible expression would be dismissed simply because older assays lacked sensitivity. Second, neuroinflammation became central to many brain-disorder models. Once diseases were analyzed through immune and glial mechanisms rather than neuron-only frameworks, CB2 became much harder to ignore. Third, receptor pharmacology itself matured. The field now thinks more in terms of efficacy, signaling bias, receptor trafficking, and context-dependent responses than simple occupancy.
That broader GPCR perspective is visible even outside the CB2 literature. The 2025 American Journal of Psychiatry article on CB1 biased signaling and schizophrenia argues that cannabinoid pharmacology should be understood through biased signaling rather than crude receptor activation. Schizophrenia affects about 24 million people worldwide, according to WHO, so this is not an academic side issue. The same logic applies to CB2. A ligand that is “CB2-selective” on paper may still produce different outcomes depending on whether it preferentially drives G-protein signaling, beta-arrestin recruitment, receptor internalization, or anti-inflammatory transcriptional programs in activated microglia.
The newer systems view reinforces this. A 2025/2026 PubMed-indexed network-analysis study identified CB1 and CB2 as highly influential nodes in the endocannabinoid system and connected receptor signaling with metabolic pathways rather than isolating receptors from the rest of cell biology. That fits what the CNS CB2 data are showing: distribution is not a fixed atlas entry. It is part of an adaptive signaling network.
The upshot is simple. CB2 should still be introduced as an immune-enriched cannabinoid receptor. But stopping there now gives the wrong picture. In the brain, CB2 is best understood as a low-basal, inducible, disease-linked receptor whose importance becomes clearest in microglia and neuroinflammatory states—and whose detection still depends heavily on method, model, and timing.
How cannabinoid receptors signal: Gi/o coupling, second messengers, and synaptic effects
Cannabinoid receptor pharmacology starts with a simple statement that becomes complicated fast: CB1 and CB2 are class A G protein-coupled receptors, and both most often signal through Gi/o proteins. That basic fact, established through foundational receptor work by Allyn Howlett and others, still holds. What has changed is the understanding of what Gi/o coupling actually means in cells. It does not mean a single downstream effect. It means a menu of possible effects whose mix depends on ligand, receptor density, phosphorylation state, membrane environment, cell type, and timing.
That distinction matters because roughly 200 million people used cannabis in 2019, or 4% of the global population aged 15 to 64 according to the World Health Organization, while the FDA states that one cannabis-derived drug product and three cannabis-related drug products are approved as of 2025. Receptor signaling is not a side issue. It is the mechanism that separates a useful antiseizure drug from sedation, a failed appetite drug from psychiatric adverse effects, and a lab-selective ligand from a clinically disappointing one.
Canonical GPCR signaling at CB1 and CB2
Canonical CB1/CB2 signaling sequence
- Ligand binding An agonist stabilizes an active receptor conformation.
- G-protein activation The receptor promotes GDP-GTP exchange on Gi/o.
- Subunit separation Galpha and Gbeta-gamma regulate downstream effectors.
- Second messenger change Adenylyl cyclase activity falls and cAMP decreases.
- Cellular effect Ion channels, transmitter release, kinases, and gene regulation shift.
In the canonical scheme, agonist binding stabilizes an active receptor conformation, the receptor acts as a guanine nucleotide exchange factor for Gi/o, Gαi/o exchanges GDP for GTP, and the Gα and Gβγ components then regulate downstream effectors. For CB1 and CB2, the classic readout is inhibition of adenylyl cyclase and a fall in intracellular cyclic AMP. That finding became one of the earliest biochemical signatures used to define cannabinoid receptor activity.
But “canonical” should not be read as “uniform.” CB1 shows high constitutive activity in several expression systems, which means the receptor can signal measurably even without agonist present. That property helps explain why inverse agonists such as rimonabant did more than block endogenous cannabinoid tone; they pushed signaling below baseline and produced marked central adverse effects. CB2 also couples to Gi/o, yet the way ligands stabilize active states differs from CB1. Structural work reviewed in Frontiers in Chemical Biology in 2026 emphasized that subtype selectivity between “CB1 and CB2” is driven by receptor-level differences that alter not just binding, but efficacy and regulation. A 2025/2026 PubMed-indexed study on subtype selectivity pushed this further by arguing that endocannabinoid selectivity is dynamic, shaped by conformational behavior rather than a fixed lock-and-key model.
This is one reason phytocannabinoids, endocannabinoids, and synthetic ligands should never be treated as interchangeable. Anandamide, identified by Raphael Mechoulam and Lumír Hanuš, and 2-arachidonoylglycerol are endogenous ligands produced on demand and rapidly inactivated. Δ9-THC is a plant-derived partial agonist with kinetics and efficacy distinct from those endocannabinoids. Synthetic agonists such as CP55,940, WIN55,212-2, or HU-210 often drive stronger receptor activation and can recruit signaling pathways to different degrees. Some ligands favor G protein signaling over β-arrestin recruitment; others do not. The American Journal of Psychiatry article in 2025 made that point directly for CB1, arguing that biased signaling is a plausible therapeutic strategy in schizophrenia, a disorder affecting about 24 million people worldwide.
CB2 adds another correction to older simplifications. It is still enriched in many immune cell populations, but the 2026 Frontiers in Behavioral Neuroscience review described “an update over the last 3 years” in which CB2 signaling gained attention in central nervous system disorders linked to neuroinflammation and neurodegeneration. So even before biased agonism enters the picture, the old binary of one “brain receptor” and one “immune receptor” already fails at the level of signaling context.
Effects on cAMP, ion channels, and neurotransmitter release
The best-known second messenger effect at both receptors is suppression of cAMP formation through inhibition of adenylyl cyclase. Lower cAMP often means lower protein kinase A activity, altered phosphorylation of downstream targets, and slower changes in gene expression through pathways such as CREB. In neurons, though, the fast effects are often more important than the slow ones.
CB1 is heavily positioned for presynaptic control. In many brain circuits it sits on axon terminals, where receptor activation reduces the probability of neurotransmitter release. This happens through a combination of Gβγ-mediated inhibition of voltage-gated calcium channels and activation of inwardly rectifying potassium conductances or other potassium currents that dampen terminal excitability. Less calcium entry means fewer vesicles fuse. The result is less transmitter released into the synaptic cleft.
That is the core mechanism behind short-range retrograde endocannabinoid signaling. A postsynaptic neuron becomes active, synthesizes endocannabinoids on demand from membrane lipid precursors, and sends them backward across the synapse to activate presynaptic CB1 receptors. The presynaptic terminal then releases less transmitter. It is a feedback brake. In excitatory synapses, that can suppress glutamate release; in inhibitory synapses, it can suppress GABA release. The direction of circuit output depends on which terminal expresses CB1. Same receptor, opposite network consequence.
| Term | What is suppressed | Mechanism described |
|---|---|---|
| DSI | Inhibition | Postsynaptic activity releases endocannabinoids that activate presynaptic CB1 and reduce GABA release |
| DSE | Excitation | Postsynaptic activity releases endocannabinoids that activate presynaptic CB1 and reduce glutamate release |
DSI and DSE Short-term forms of endocannabinoid-mediated synaptic plasticity in which postsynaptic depolarization suppresses inhibitory transmission (DSI) or excitatory transmission (DSE) through presynaptic CB1 activation.
Classic physiological terms capture this: depolarization-induced suppression of inhibition, DSI, and depolarization-induced suppression of excitation, DSE. Both are short-term forms of synaptic plasticity driven by endocannabinoid release and presynaptic CB1 activation. Longer-lasting effects also occur, including endocannabinoid-mediated long-term depression at some synapses. Those phenomena matter because they connect receptor biochemistry to behavior: pain processing, fear extinction, habit learning, appetite, motor control, and seizure threshold all depend on this tuning of release probability.
The details are not trivial. A partial agonist like Δ9-THC may not mimic the full pattern produced by a brief endogenous burst of 2-AG. Nor will a synthetic full agonist necessarily preserve physiological timing. Dose matters. So does receptor reserve. In a synapse with dense CB1 expression, even a partial agonist can produce a large effect on transmitter release. In a different tissue, the same ligand may look weak.
CB2 has less established direct synaptic physiology than CB1, but it also reduces cAMP and can regulate calcium signaling, kinase pathways, and inflammatory mediator release in immune and glial cells. That makes CB2 relevant to neuron-glia communication, especially in disease states where receptor expression changes. The recent network-analysis paper indexed in PubMed in 2025/2026 treated CB1 and CB2 as influential nodes in broader endocannabinoid and metabolic signaling, which is a better framing than treating them as isolated switches.
Desensitization, internalization, and receptor regulation
How receptors adapt to repeated agonist exposure
- Phosphorylation Intracellular receptor regions are modified by GPCR kinases and other kinases.
- Beta-arrestin recruitment Arrestins uncouple receptors from G proteins and can start additional signaling.
- Desensitization The receptor becomes less responsive.
- Internalization Receptors are pulled into endocytic pathways.
- Fate after uptake Receptors may recycle back to the surface or be degraded.
No receptor can be activated continuously without consequences. For CB1 and CB2, prolonged or repeated agonist exposure commonly leads to phosphorylation of the receptor’s intracellular regions by GPCR kinases and other kinases, recruitment of β-arrestins, uncoupling from G proteins, and then internalization through endocytic pathways. Desensitization comes first. Endocytosis often follows. Recycling or degradation comes after that.
For CB1, this regulatory cycle is a major reason that acute and chronic effects differ. Strong agonists can trigger rapid desensitization in cell systems and measurable tolerance in vivo. Region-specific regulation matters here. CB1 receptors do not desensitize equally in all neuronal populations, which helps explain why tolerance develops unevenly across cannabinoid effects. Analgesic responses, hypothermia, memory disruption, and motor effects can shift at different rates because the receptor is regulated differently across circuits.
β-arrestins are not only off-switches. They can scaffold their own signaling cascades, including MAP kinase pathways, which is why arrestin recruitment has become central to biased agonism. A ligand that strongly inhibits cAMP but weakly recruits β-arrestin may behave differently from one that does both efficiently. That is not a theoretical fine point anymore; it is a drug-design strategy. The 2025 American Journal of Psychiatry discussion of CB1 bias in schizophrenia reflects a broader GPCR lesson: avoiding certain signaling branches may reduce some liabilities, but selectivity for a pathway does not guarantee clinical success.
Internalization itself is also ligand-dependent. Some agonists drive extensive receptor endocytosis; others produce limited internalization despite G protein activation. Allosteric modulators complicate the picture further by changing how orthosteric ligands stabilize receptor states. This is where structural pharmacology meets therapeutics. The 2026 structural review made clear that receptor conformation controls signaling efficacy and receptor regulation together, not as separate topics.
That is the key signaling lesson to carry forward. CB1 and CB2 are not simple on-off detectors for cannabinoids. They are regulated hubs whose output changes across milliseconds to days. Any serious attempt to target them, whether for epilepsy, pain, psychosis, or inflammatory disease, has to account for Gi/o coupling, second messengers, ion-channel control, synaptic location, and the fact that the receptor will adapt to being pushed.
Biased signaling: why one receptor can produce different biological outcomes
The old picture of cannabinoid pharmacology treated a receptor like a light switch: agonists turn it on, antagonists turn it off, and everything else follows from where that receptor happens to be expressed. That picture is not adequate for CB1 or CB2. It misses why two ligands acting at the same receptor can produce sharply different behavioral, cognitive, inflammatory, or therapeutic effects. It also misses why drug discovery around cannabinoid receptors has repeatedly produced compounds that looked promising in vitro yet proved disappointing, intolerable, or clinically ambiguous.
That matters well beyond academic receptor theory. The World Health Organization estimated that 200 million people used cannabis in 2019, about 4% of the global population aged 15–64. Schizophrenia affects about 24 million people worldwide. Against that background, the pharmacology of CB1 is not a niche issue. It sits at the intersection of public health, psychiatry, and drug design. The U.S. FDA, as of 2025, had approved one cannabis-derived drug product and three cannabis-related drug products, a small number compared with the scale of clinical interest. One reason progress has been slower than public discussion suggests is that cannabinoid receptor signaling is not simple receptor occupancy. It is pathway selection.
What biased agonism means in GPCR pharmacology
CB1 and CB2 are class A G protein-coupled receptors. Foundational work by Allyn Howlett established CB1 as a Gi/o-coupled cannabinoid receptor, helping move the field from vague pharmacology to receptor-defined mechanisms. But Gi/o coupling is only the start of the story. Once a ligand binds, the receptor can adopt more than one active shape, and those shapes do not signal identically. Some receptor conformations favor G protein activation. Others more strongly recruit beta-arrestins. Some states promote receptor phosphorylation, desensitization, or internalization. Others produce longer-lived signaling from the plasma membrane or from endosomal compartments.
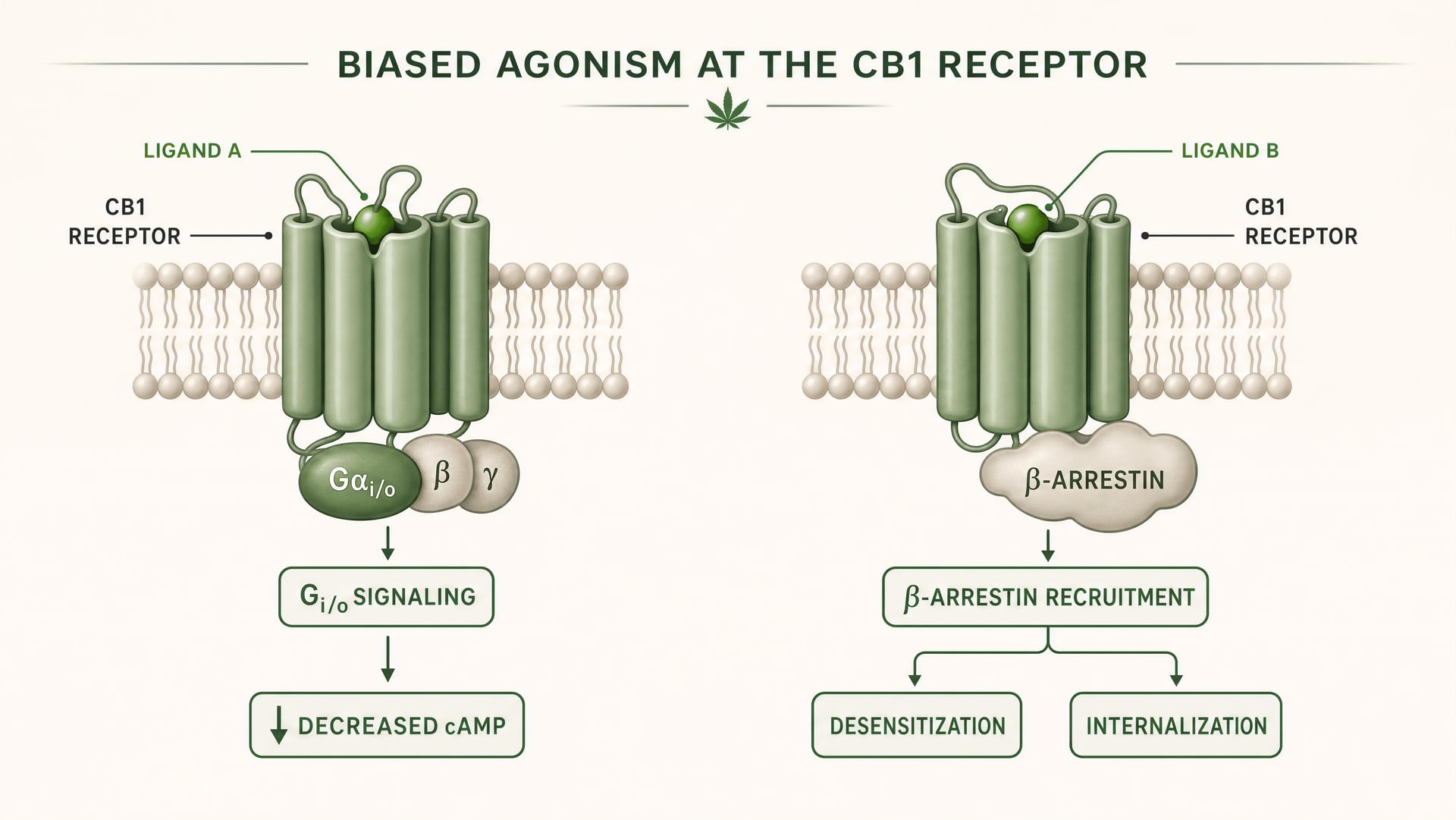
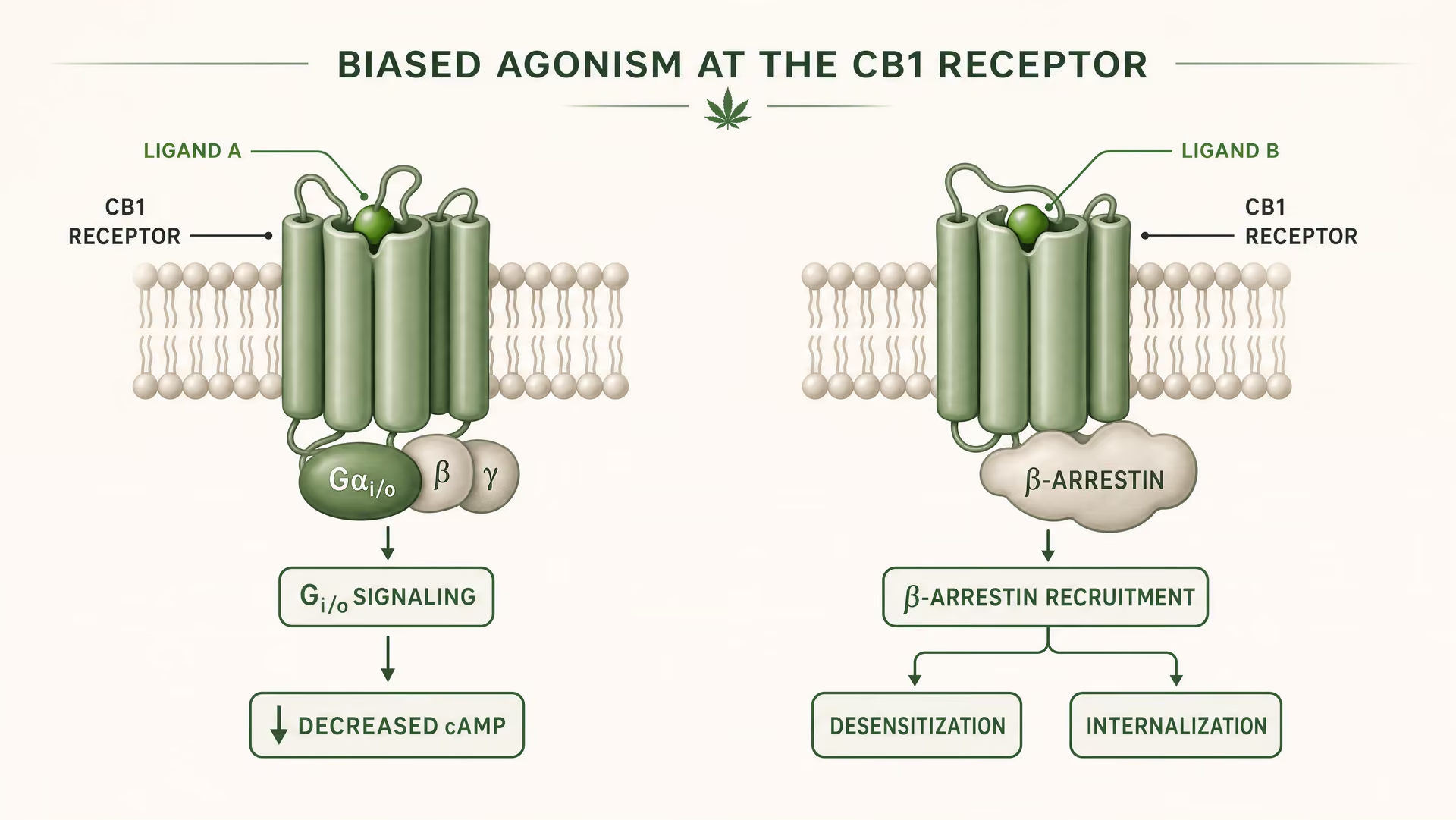{kind=link}
Biased agonism A property of a ligand that favors some downstream signaling pathways over others at the same receptor.
That is biased agonism in plain terms: different ligands stabilize different receptor conformations, and those conformations favor different downstream pathways. A receptor is not merely on or off. It is conformationally directed.
For CB1, this is especially important because the receptor sits in a signaling environment that is dense, plastic, and highly cell-type dependent. In a cortical glutamatergic terminal, a ligand may reduce transmitter release through Gi/o-mediated inhibition of adenylyl cyclase and modulation of ion channels. In a GABAergic interneuron, the same receptor can shift local circuit balance in a very different direction. If the ligand also promotes strong beta-arrestin recruitment, the receptor may internalize faster, shortening one effect while opening another. Timing changes. Signal location changes. The physiological readout changes.
This is not theoretical hair-splitting. The 2026 Frontiers in Chemical Biology structural review on cannabinoid receptors makes the point clearly: ligand selectivity at CB1 and CB2 depends on receptor-level structural differences that alter binding, signaling efficacy, and receptor regulation. The key word there is regulation. A ligand may be similar in affinity yet differ in efficacy, arrestin recruitment, residence time, or propensity to trigger desensitization. The 2025/2026 PubMed-indexed study on the dynamic mechanism of subtype selectivity pushes the same idea further by arguing that selectivity emerges from conformational dynamics, not a static lock-and-key model. Endocannabinoids, phytocannabinoids, and synthetic ligands should therefore not be lumped together. Anandamide, discovered by Raphael Mechoulam and Lumír Hanuš, does not behave like delta-9-tetrahydrocannabinol, and neither behaves like a highly optimized synthetic probe.
Biased signaling also explains why allosteric modulators attract so much interest. An allosteric ligand may not directly activate CB1 the way an orthosteric agonist does, yet it can reshape the receptor’s signaling preferences, amplifying one pathway and damping another. That opens a route to fine control. In principle.
CB1 biased signaling as a schizophrenia research direction
The 2025 American Journal of Psychiatry article makes the strongest recent case that CB1 biased signaling is not just a pharmacology concept but a plausible therapeutic strategy for schizophrenia. That argument deserves attention because schizophrenia research has usually approached cannabinoids through epidemiology, risk association, or broad warnings about psychosis. The AJP paper shifts the frame. It asks whether the problem is not “cannabinoids” in general, or even “CB1 activation” in general, but which CB1 signaling states are being engaged, in which circuits, and for how long.
That is a much better question.
CB1 is one of the most abundant GPCRs in the brain, with heavy expression in cortex, hippocampus, basal ganglia, and cerebellum, but abundance alone does not explain clinical effects. Schizophrenia involves dysregulated salience, cognition, perception, and network coordination across cortical and subcortical systems. A receptor positioned to tune glutamate, GABA, and dopamine-related circuit activity is therefore relevant by design. The AJP article argues that biased CB1 ligands might separate therapeutically useful circuit effects from liabilities such as cognitive impairment, anxiety, dysphoria, or psychotomimetic responses.
That is an ambitious claim, but it is not speculative hand-waving. It follows the broader GPCR field, where pathway bias has already changed how researchers think about opioid, angiotensin, and dopamine receptor drugs. The translational hope at CB1 is that certain signaling outputs may improve cortical network function or damp aberrant circuit states without reproducing the full adverse profile associated with high-efficacy CB1 agonism.
Schizophrenia is a good test case because the clinical bar is high. A candidate drug cannot merely alter behavior in a rodent assay. It must avoid worsening psychosis, sedation, and cognitive disruption in people already vulnerable to those problems. That immediately makes pathway selectivity more than a medicinal chemistry preference. It becomes a safety requirement.
The AJP framing also helps correct a common oversimplification in cannabis discussions. Delta-9-THC is a phytocannabinoid with partial agonist activity at CB1, but its effects reflect dose, timing, receptor reserve, local endocannabinoid tone, and pathway engagement across different neuronal populations. A synthetic CB1 ligand built to favor one intracellular route could look very different from THC even if both “hit CB1.” The same applies in reverse: two compounds that both improve a preclinical schizophrenia-relevant endpoint might diverge badly on cognition or affect if one drives arrestin-heavy signaling and the other does not. Receptor identity alone cannot predict the whole phenotype.
Why pathway selectivity matters for safety and efficacy
Pathway selectivity matters because efficacy is not a single dimension. A cannabinoid drug can be potent and still clinically poor. It can be selective for CB1 and still fail. It can avoid CB2 entirely and still produce unwanted immune or metabolic effects through network crosstalk. The 2025/2026 integrative network analysis indexed in PubMed identified CB1 and CB2 as highly influential nodes in the endocannabinoid system and mapped their signaling onto metabolic pathways. That systems view is essential. Receptors do not operate in isolation, and pathway bias at one node can ripple into broader physiological programs.
For CB1, safety concerns are obvious. Strong central CB1 activation can produce memory impairment, altered perception, anxiety, tachycardia, and in susceptible individuals, psychosis-related effects. Any therapeutic program aimed at pain, appetite, mood, addiction, or schizophrenia has to confront that liability profile. A ligand that preserves a desired Gi/o-mediated synaptic effect while limiting beta-arrestin-driven desensitization or other adverse signaling cascades could, in theory, widen the therapeutic window. But “in theory” matters. Many biased-ligand programs in GPCR pharmacology have shown that bias measured in one assay system does not always predict outcomes in vivo. Cell background, receptor density, effector expression, and kinetics can all shift the apparent bias.
CB2 offers a cautionary parallel. The 2026 Frontiers in Behavioral Neuroscience review describes an update over the last 3 years in which CB2 signaling gained attention in central nervous system disorders through links to neuroinflammatory and neurodegenerative mechanisms. That directly undercuts the old idea that CB2 is irrelevant to the brain. Even so, simply targeting CB2 does not guarantee a useful anti-inflammatory medicine. Distribution is more graded than the old brain-versus-body split, and signaling consequences still depend on ligand and context.
So the practical lesson is blunt: receptor subtype selectivity is necessary, but not sufficient. Pathway selectivity may be the difference between a cannabinoid that looks therapeutic, one that is intoxicating, and one that fails in trials because it cannot separate benefit from adverse effect. For CB1, especially in psychiatry, that distinction is likely to decide whether the receptor remains a cautionary tale or becomes a viable drug target.
Structural biology of CB1 and CB2: how shape drives selectivity
Structural biology changed the way cannabinoid receptors are discussed. The old shortcut—CB1 explains intoxication, CB2 explains inflammation—misses the fact that both receptors are class A G protein-coupled receptors whose behavior depends on shape, motion, binding depth, and the signaling partners available in a given cell. That matters far beyond basic pharmacology. Cannabis was used by an estimated 200 million people in 2019, or 4% of the global population aged 15–64, according to the WHO, yet the FDA still lists only one cannabis-derived drug product and three cannabis-related drug products as approved in 2025. One reason for that gap is structural: it is hard to make cannabinoid ligands that hit the right receptor, in the right way, for the right duration.
The Frontiers in Chemical Biology review from 2026 makes this point clearly. CB1 and CB2 do not just differ in where they are expressed. They differ in the architecture of their ligand-binding pockets, the shape and flexibility of their extracellular loops, the packing of their transmembrane helices, and the conformational states they prefer after a ligand binds. Those features influence not only selectivity, but also efficacy, desensitization, internalization, and pathway bias.
What structural studies reveal about receptor pockets
An orthosteric pocket is the main binding cavity where endogenous ligands such as anandamide and 2-arachidonoylglycerol, phytocannabinoids such as THC, and many synthetic ligands make their primary contact. In CB1 and CB2, that pocket sits within the bundle of seven transmembrane helices, capped in part by extracellular loops that can either open access or restrict it.
Cryo-EM and X-ray structures over the last several years showed that cannabinoid receptors do not behave like rigid locks waiting for a key. They are better understood as moving targets with preferred shapes. The 2026 Frontiers in Chemical Biology review emphasizes that the orthosteric cavities of CB1 and CB2 are similar enough to bind overlapping classes of ligands, yet different enough in size, residue identity, and local flexibility to shift affinity and signaling outcome. That is why closely related compounds can separate pharmacologically. A small change in substituent bulk, polarity, or tail length can alter how deeply a ligand penetrates the pocket, which helices it pushes, and whether the receptor settles into a G protein-favoring or arrestin-favoring state.
CB1 has been especially informative structurally because many high-resolution inactive and active-state models now exist. One recurring theme is that its pocket is expansive and hydrophobic, fitting the lipophilic nature of many cannabinoids. Extracellular loop 2 and the upper parts of several helices help shape the entryway. Transmembrane helices are the seven membrane-spanning segments that make up the receptor core; when a ligand binds, these helices can shift relative to each other. The most pharmacologically important motion is usually at the intracellular side, where outward movement of helix 6 helps create a docking site for Gi/o proteins. That shift is one hallmark of receptor activation.
CB2 shares the same overall GPCR fold, but the Frontiers review argues that subtype-specific amino acid differences around the pocket and loop regions give medicinal chemists exploitable handles for selectivity. The point is not that one pocket is simply “brain-like” and the other “immune-like.” The point is geometric and energetic. Distinct residues alter cavity contour, local hydrogen-bonding options, aromatic stacking, and the flexibility of access channels through which ligands enter from the membrane.
A 2025/2026 PubMed-indexed study on the dynamic mechanism of subtype selectivity pushed this further by arguing that endocannabinoid selectivity is not just a matter of static binding affinity. Conformational dynamics matter. In plain terms, a receptor can sample multiple shapes before and after ligand binding, and some ligands stabilize a selective shape better than others. That helps explain why endogenous lipids, phytocannabinoids, and synthetic ligands can show different subtype preferences even when their scaffolds look related on paper.
Determinants of ligand selectivity between CB1 and CB2
Selectivity starts with contact chemistry but does not end there. The Frontiers in Chemical Biology review frames selectivity as the product of receptor-level structural differences that affect binding, signaling efficacy, and regulation all at once. That is the right frame. A ligand may be CB2-selective in a radioligand binding assay yet lose its practical advantage if it also drives receptor states that cause rapid tolerance or weak signaling in disease-relevant cells.
Several structural features repeatedly come up. First, the amino acid composition of the orthosteric pocket differs enough between CB1 and CB2 to change how a ligand’s head group, core, and hydrophobic tail are accommodated. Second, extracellular loops help shape entry and orientation. Third, the upper and middle regions of transmembrane helices can bias the receptor toward slightly different active-state ensembles. A conformational state is simply one of the receptor’s possible shapes at a given moment. Different ligands do not just bind to a receptor; they stabilize one subset of those shapes.
That is why subtype selectivity is often modest for natural cannabinoids. THC, for example, engages both receptors. Anandamide and 2-AG also act at both, though with context-dependent differences in potency, efficacy, and metabolism. Synthetic ligands have been more useful for teasing apart structure-selectivity relationships because chemists can systematically alter features such as side-chain length, ring constraints, and polar substituents. Even then, clean separation is hard. CB1 and CB2 are homologous enough that a compound designed for one often retains meaningful activity at the other.
This has practical consequences. Drug developers have long chased CB2-selective agonists in the hope of getting anti-inflammatory or analgesic effects without strong CB1-mediated central adverse effects. Sometimes that strategy works pharmacologically, but it is not a free pass. The 2026 Frontiers in Behavioral Neuroscience review stresses that CB2 has drawn attention in central nervous system disorders over “the last 3 years,” undermining the simple view that CB2 is irrelevant to the brain. So even a “peripheral” CB2 ligand cannot be interpreted using outdated receptor maps.
Why efficacy and regulation are also structural questions
Potency answers how much ligand is needed. Efficacy asks what that ligand makes the receptor do once bound. Structural biology links the two but does not confuse them. Two ligands can occupy the same pocket and produce very different responses because they stabilize different active conformations.
For CB1, this is central to current therapeutic thinking. The American Journal of Psychiatry article from 2025 argues that biased signaling at CB1 may be a plausible strategy for schizophrenia, a disorder that affects about 24 million people worldwide. Biased agonism means a ligand favors one downstream pathway over another, often Gi/o signaling over beta-arrestin recruitment, or vice versa. Structurally, that bias arises because the ligand shifts the receptor’s intracellular face into a shape that better fits one signaling partner than another. That is not an abstract idea. It is a medicinal chemistry target.
Beta-arrestins matter because they contribute to desensitization and internalization. Desensitization means the receptor becomes less responsive after repeated or sustained activation. Internalization means the receptor is pulled into the cell from the surface membrane. Both processes are influenced by receptor conformation. Some ligands strongly activate G proteins but only weakly recruit arrestins; others do both. A behaviorally tolerable CB1 drug may need exactly this sort of separation, rather than simple blockade or full agonism.
Allosteric modulation adds another layer. An allosteric ligand binds outside the orthosteric pocket and changes how the receptor responds to other ligands. Structurally, this is appealing because it may allow finer control over receptor shape than blasting the main pocket with a high-efficacy agonist. For cannabinoid pharmacology, that could mean preserving useful signaling while reducing adverse effects tied to excessive CB1 activation.
The larger lesson is blunt. Receptor shape is not a side detail. It is the explanation for why phytocannabinoids, endocannabinoids, and synthetic compounds can produce different clinical profiles even when they target the same receptor family. Since CB1 and CB2 sit at influential nodes in endocannabinoid-system network analyses, as a 2025/2026 PubMed-indexed systems study reported, errors in selectivity or efficacy can propagate across many pathways. Structural insight does not guarantee a successful drug. It does explain why selectivity, potency, and tolerability remain so hard to align.
Endocannabinoids and subtype selectivity: anandamide, 2-AG, and dynamic receptor behavior
Endocannabinoid pharmacology is often explained with a shortcut that sounds tidy but misleads: anandamide and 2-arachidonoylglycerol (2-AG) are treated as if they were the body’s built-in “standard agonists,” useful mainly as reference points for what CB1 and CB2 normally do. That framing misses the central fact. These are not stable, freely circulating hormones that encounter receptors under uniform conditions. They are short-lived lipid mediators made on demand, in membranes, by cell-specific enzyme systems, then destroyed quickly. Their apparent receptor preference can shift with membrane composition, receptor conformations, local enzyme activity, and the timing of release.
That matters beyond academic receptor theory. Cannabis was used by an estimated 200 million people in 2019, or 4% of the global population aged 15–64, according to WHO, and cannabinoid-targeting medicines are already in the clinic: the FDA said in 2025 that it has approved one cannabis-derived drug product and three cannabis-related drug products. If endogenous ligands do not behave like simple textbook controls, then comparing phytocannabinoids or drug candidates against them is not straightforward.
Why endogenous ligands are not simple reference compounds
Anandamide, identified in 1992 by Devane, Hanus, Breuer, Mechoulam, and colleagues, and 2-AG, established a few years later as another major endocannabinoid, differ in more than potency tables. They come from different biosynthetic routes, reach receptors on different time scales, and are cleared by different enzymes. Anandamide is generated mainly from N-arachidonoyl phosphatidylethanolamine through pathways involving NAPE-PLD, although alternative routes exist. 2-AG is produced largely from diacylglycerol by DAGLα and DAGLβ. Those are not minor biochemical footnotes; they determine where and when receptor activation happens.
Even the older claim that anandamide is a CB1-preferring ligand while 2-AG is a full agonist at both CB1 and CB2 can become too blunt when moved from purified systems to living tissue. Anandamide is also a substrate for fatty acid amide hydrolase, or FAAH, and can engage non-cannabinoid targets such as TRPV1 under some conditions. 2-AG is present in brain tissue at much higher basal levels than anandamide in many measurements and is hydrolyzed mainly by monoacylglycerol lipase, or MAGL, with ABHD6 and ABHD12 also contributing. So the receptor “input” each ligand provides is not just a matter of affinity or efficacy at CB1 versus CB2. It is a matter of whether the ligand is made at the synapse, inside an immune cell, near an intracellular membrane, or in a compartment rich in degradative enzymes.
Allyn Howlett’s receptor pharmacology work helped establish CB1 as a bona fide GPCR coupled largely to Gi/o proteins. That framework remains essential, but it no longer supports a simple endogenous-reference-drug model. CB1 and CB2 can signal through inhibition of adenylyl cyclase, regulation of ion channels, MAP kinase pathways, beta-arrestin recruitment, receptor internalization, and desensitization. A ligand that looks “weaker” in one assay may produce longer signaling in a tissue where degradation is slow, or may favor one downstream pathway over another. The 2025 American Journal of Psychiatry article on CB1 biased signaling makes this point in a disease setting: the therapeutic question in disorders such as schizophrenia, which affects about 24 million people worldwide, is not merely whether CB1 is activated, but how.
Dynamic mechanisms of subtype selectivity
The recent subtype-selectivity study indexed in PubMed (PMID: 41962866) is important because it pushes the field away from a lock-and-key story. Its core message is that endocannabinoid selectivity can arise from dynamic mechanisms. In other words, receptor subtype preference is not explained only by a fixed difference in how tightly a ligand binds CB1 versus CB2. The ligand-receptor complex samples multiple conformational states, and those states differ between CB1 and CB2.
That finding fits with the 2026 Frontiers in Chemical Biology review, which argues that structural differences between CB1 and CB2 shape binding, signaling efficacy, and receptor regulation. The receptors share substantial sequence similarity, yet small differences in orthosteric pocket architecture, extracellular loops, transmembrane helix movements, and intracellular coupling surfaces can bias what happens after ligand engagement. A ligand may enter similar pockets in both receptors but stabilize a more signaling-competent state in one subtype, or favor different transitions between inactive, intermediate, and active states.
For endocannabinoids, this is especially plausible because they are flexible lipids rather than rigid synthetic scaffolds. Flexibility is a pharmacological variable. Anandamide can adopt multiple conformations, and 2-AG is similarly dynamic in membranes. The question then becomes not only “Does it bind?” but “Which receptor states does it favor, how long are those states occupied, and what proteins are available to couple next?” A CB1-rich neuron and a CB2-expressing microglial cell do not present the same signaling environment, even before differences in receptor density are considered.
This dynamic view also weakens the old assumption that endogenous ligands define a single physiological baseline against which THC, cannabidiol, or synthetic cannabinoids can be ranked. THC is a phytocannabinoid with its own efficacy pattern and kinetic behavior. Endocannabinoids are event-driven signals, often phasic and local. A synthetic CB2 agonist may look selective in a recombinant assay yet produce unexpected results in tissue if receptor reserve, membrane cholesterol, heteromer formation, or beta-arrestin handling differ. Selectivity on paper is not destiny.
How local synthesis and degradation shape receptor engagement
In vivo, endocannabinoid signaling is governed as much by enzymology as by receptor pharmacology. At many central synapses, 2-AG is produced postsynaptically in response to calcium rises or Gq/11-coupled receptor activation, then travels retrogradely to presynaptic CB1 receptors and suppresses neurotransmitter release. This is a timing mechanism measured in seconds, not a diffuse background tone. The signal ends when MAGL and related hydrolases remove 2-AG. Anandamide often behaves differently, with lower tissue abundance, different release conditions, and stronger sensitivity to FAAH-controlled breakdown.
That means changing an enzyme can alter receptor engagement without changing the receptor at all. FAAH inhibition raises anandamide tone, but the physiological result depends on where anandamide is produced, whether TRPV1 is also present, and whether CB1 receptors in that microdomain are desensitized or internalized. MAGL inhibition can markedly elevate 2-AG, but chronic elevation may drive CB1 desensitization in some systems. More ligand does not always mean more useful signaling.
The systems-biology study indexed as PMID: 42129940 reinforces this broader picture by placing CB1 and CB2 among influential nodes connected to metabolic pathways, not isolated switches. That is the right scale for understanding endocannabinoid action. Ligand availability, membrane access, receptor conformation, G-protein coupling, beta-arrestin recruitment, and degradation all interact. So do tissue gradients. CB2 is not simply “outside the brain,” and CB1 is not only “the psychoactivity receptor.” Endocannabinoid signaling is local, conditional, and kinetic. Any account of subtype selectivity that ignores those features will misread both physiology and drug development.
The endocannabinoid system as a network, not a two-receptor diagram
The endocannabinoid system makes little sense if it is drawn as two circles labeled CB1 and CB2 with arrows pointing outward. That cartoon helped the field early on, especially after Allyn Howlett’s receptor pharmacology work and the identification of anandamide by Raphael Mechoulam and Lumír Hanuš, but it now hides more than it reveals. The practical stakes are large. The World Health Organization estimated that 200 million people used cannabis in 2019, about 4% of the global population aged 15–64, while the U.S. FDA states that one cannabis-derived drug product and three cannabis-related drug products are approved as of 2025. A receptor system that touches so many exposures and so many therapeutic claims cannot be treated as a simple brain receptor plus immune receptor pair.
A systems view starts from a basic fact: endocannabinoids are lipids made on demand, degraded quickly, and embedded in the same biochemical space as inflammatory mediators, membrane remodeling pathways, and synaptic feedback loops. Anandamide and 2-arachidonoylglycerol are not isolated “messages.” They are produced from membrane precursors, compete with other lipid pathways for substrates, and act in tissues where receptor density, cell type, enzyme expression, and signaling partners all differ. That is why the same ligand can look anxiolytic in one setting, intoxicating in another, anti-inflammatory somewhere else, and clinically underwhelming in a trial.
What integrative network analysis adds
The PubMed-indexed integrative network analysis study published in 2025/2026 (PMID: 42129940) is useful because it shifts attention from single targets to system organization. Instead of asking only which ligand binds CB1 or CB2, the authors map the endocannabinoid system as an interaction network that includes receptors, biosynthetic and degradative enzymes, lipid intermediates, and pathway-level links to inflammation, metabolism, and neuronal signaling. In that framework, influence is not defined just by receptor abundance. It is defined by connectivity and by how perturbations spread through the network.
That matters for interpretation. If a drug changes CB1 signaling, the downstream effect is not limited to Gi/o-mediated inhibition of adenylyl cyclase. It may alter neurotransmitter release, shift calcium and potassium channel behavior, change beta-arrestin recruitment, trigger receptor internalization, and indirectly modify the flux of lipid substrates that also feed eicosanoid and other inflammatory pathways. A network model captures these cross-system consequences better than a receptor occupancy chart.
This is also where the old “CB1 in brain, CB2 in immune cells” shorthand breaks down. Distribution is graded, cell-specific, and state-dependent. CB1 is abundant in many neuronal populations, but not uniformly across the brain, and it also appears in peripheral tissues relevant to energy balance, nociception, and organ function. CB2 is enriched in immune lineages, yet newer central nervous system work has pushed well past the idea that it is irrelevant to brain biology. A 2026 Frontiers in Behavioral Neuroscience review described this as “an update over the last 3 years,” emphasizing CB2 signaling in neuroinflammatory and neurodegenerative settings. The point is not that CB2 suddenly became “the brain receptor too.” The point is that receptor meaning depends on which cells are active, injured, inflamed, or adapting.
Network analysis helps explain why receptor-targeted drugs often produce broader effects than expected. A CB1-active phytocannabinoid such as delta-9-THC, an endocannabinoid such as anandamide, and a synthetic agonist can all engage the same receptor while driving different temporal and spatial outcomes. Ligand class matters. So do efficacy, residence time, metabolism, and access to different receptor conformations.
CB1 and CB2 as influential nodes
The integrative network study identifies CB1 and CB2 as highly influential nodes, not because they operate alone, but because they sit at control points where many pathways intersect. In network terms, influential nodes are places where local changes can propagate widely. In pharmacology terms, they are receptors whose activation state can redirect synaptic signaling, immune responses, and cellular stress programs at the same time.
CB1 is the clearer example of why “influential” does not mean “simple.” It signals mainly through Gi/o proteins, but that is only the start. Depending on ligand and context, CB1 can engage beta-arrestins, undergo phosphorylation-dependent desensitization, internalize, recycle, or remain persistently active at the membrane. The American Journal of Psychiatry article from 2025 argues that biased signaling at CB1 is a plausible therapeutic strategy for schizophrenia, a disorder that the WHO says affects about 24 million people worldwide. That proposal only makes sense if CB1 is understood as a signaling hub with separable outputs, not an on/off switch for intoxication. A ligand that favors one signaling branch over another may preserve some therapeutic effects while reducing adverse ones. May, not will. Bias can reduce liabilities, but it does not erase the fact that CB1 sits inside circuits governing cognition, reward, motor control, pain processing, and appetite.
CB2 shows a similar problem from the opposite direction. It was long cast as the safer target because of lower association with acute psychoactive effects. That framing is too optimistic. CB2-directed ligands can still produce disappointing or mixed outcomes if disease-relevant signaling depends on cell state, receptor inducibility, inflammatory tone, or cross-talk with metabolic enzymes. The recent structural review in Frontiers in Chemical Biology, published in 2026, emphasizes that selectivity between “CB1 and CB2” depends on receptor-level structural differences that change binding, efficacy, and receptor regulation. The 2025/2026 PubMed-indexed study on subtype selectivity (PMID: 41962866) pushes this further by arguing that selectivity is dynamic, shaped by conformational behavior rather than a static lock-and-key fit. That is a strong reason to stop talking about CB1-selective or CB2-selective compounds as if selectivity were a fixed property with fixed biological consequences.
Links between receptor signaling and metabolic pathways
The most valuable contribution of the network perspective is that it reconnects receptor pharmacology with lipid metabolism. Endocannabinoid signaling is built from arachidonic-acid-related chemistry. Anandamide and 2-AG are generated and degraded by enzyme systems that also influence the availability of bioactive lipids involved in inflammation and tissue response. A drug that blocks FAAH or MAGL does not merely “raise endocannabinoids.” It redistributes signaling traffic across a metabolic web.
That can be useful. It can also backfire. Elevating 2-AG, for example, may enhance CB1 or CB2 signaling in some compartments while altering prostaglandin-related pathways in others through substrate diversion or downstream oxidation. The clinical implication is straightforward: receptor-targeted and enzyme-targeted strategies can have system-wide consequences even when their design goal is narrow. This is one reason selective compounds have not automatically translated into clean clinical profiles in pain, psychiatric disease, or inflammatory disorders, despite the high unmet need. Chronic pain alone affects nearly 1 in 5 adults in the United States, according to the National Academies, yet cannabinoid pharmacology has not yielded a universally reliable receptor-based solution.
Seen this way, CB1 and CB2 are better understood as control nodes embedded in lipid, immune, and synaptic circuits. Their druggability is real. So is their capacity to surprise.
CB1 as a drug target: promise, liabilities, and the problem of central side effects
CB1 remains one of the most tempting targets in neuropharmacology because it sits at a strategic control point in synaptic signaling. Allyn Howlett’s receptor pharmacology work established that cannabinoid effects were not a vague membrane phenomenon but receptor-mediated biology, and the later discovery of anandamide by Raphael Mechoulam and Lumír Hanuš gave that receptor an endogenous signaling system. That history matters. A receptor that helps tune neurotransmitter release across pain circuits, feeding circuits, stress pathways, and reward networks looks like a plausible handle for several difficult disorders at once. It also creates a trap: when one receptor is embedded in so many core brain functions, broad manipulation rarely stays confined to the desired effect.
That problem is not academic. The World Health Organization estimates that 200 million people used cannabis in 2019, about 4% of the global population aged 15–64, so CB1 pharmacology is already operating at population scale through plant cannabinoids, especially delta-9-THC. Yet approved cannabinoid medicines remain few: the U.S. FDA states that it has approved one cannabis-derived drug product and three cannabis-related drug products as of 2025. The gap between widespread exposure and limited approved therapeutics says something important about CB1. The biology is compelling. The drugging is hard.
Therapeutic rationale in pain, appetite, and neuropsychiatry
The attraction starts with where CB1 acts and what it does. CB1 is highly expressed in many presynaptic terminals in the central nervous system, where endocannabinoids are often produced on demand in postsynaptic cells and travel backward across the synapse to suppress transmitter release. This retrograde system can dampen glutamate, GABA, and other signaling streams depending on cell type and circuit. For pain, that is an obvious rationale: if nearly 1 in 5 adults in the United States live with chronic pain, as the National Academies noted in 2017, a receptor that can reduce nociceptive transmission and alter pain affect is bound to attract attention.
But CB1 is not a simple analgesic switch. In some circuits, suppressing inhibitory GABA release can disinhibit downstream neurons rather than quiet them. In others, glutamatergic suppression dominates. The effect depends on the synapse, the state of the network, and the ligand. Endocannabinoids such as anandamide and 2-AG are brief, locally generated signals. Phytocannabinoids and synthetic agonists arrive with different kinetics, different efficacy, and often much broader receptor engagement over time. That distinction helps explain why “CB1 activation reduces pain” is too crude to guide safe clinical design.
Appetite is a second major rationale. CB1 signaling promotes food intake and interacts with hypothalamic and mesolimbic circuitry that links energy homeostasis to reward. This made CB1 antagonism an appealing anti-obesity strategy and CB1 agonism or indirect enhancement a potential route for cachexia or appetite loss. The logic was not irrational. It was rooted in basic physiology. Yet appetite is inseparable from mood, salience, and stress processing, all of which are also shaped by CB1.
Neuropsychiatry is where the receptor becomes most interesting and most dangerous. CB1 modulates dopamine-relevant circuits, cortical inhibition, hippocampal plasticity, fear learning, and stress responsivity. That gives it theoretical relevance to anxiety disorders, trauma-related conditions, depression, substance use disorders, and psychotic illness. The American Journal of Psychiatry argued in 2025 that biased CB1 signaling is a plausible strategy for schizophrenia, a disorder that affects about 24 million people worldwide according to WHO. The significance of that paper is not that a CB1 drug for schizophrenia is ready. It is that the field has moved beyond crude agonism and antagonism toward pathway-selective pharmacology, borrowing from the broader GPCR playbook.
Why CB1 targeting is scientifically attractive but clinically difficult
CB1 is scientifically attractive for exactly the same reason it is clinically difficult: it is a dense signaling hub. At the canonical level, CB1 couples primarily to Gi/o proteins, reducing adenylyl cyclase activity, modulating ion channels, and suppressing neurotransmitter release. That alone would be enough to make it important. But it also recruits beta-arrestins, undergoes desensitization and internalization, and can be shaped by allosteric modulators and ligand-specific conformational states. The 2026 Frontiers in Chemical Biology structural review makes this point clearly for both CB1 and CB2: ligand selectivity and efficacy arise from receptor-level structural differences that affect binding, signaling output, and receptor regulation. Similar-looking cannabinoids can therefore produce meaningfully different pharmacology.
That is not just a medicinal chemistry detail. It goes straight to side effects. A ligand that strongly drives one active conformation may produce more receptor internalization, more tolerance, or a different behavioral profile than a ligand that stabilizes another state. A 2025/2026 PubMed-indexed study on subtype selectivity further argued that endocannabinoid selectivity is dynamic, not a fixed lock-and-key event, reinforcing the idea that receptor behavior changes with conformational context. Drug developers do not face a single CB1 target. They face a moving ensemble.
This is why blunt CB1 agonists and antagonists have disappointed so often. Strong central agonism can impair memory, attention, psychomotor performance, and anxiety regulation while also engaging reward pathways in ways that complicate dependence liability. Strong central antagonism can reduce appetite, yes, but it can also interfere with the same stress-buffering and affective circuits that endogenous cannabinoids normally stabilize. CB1 is deeply involved in cognition, reward, and stress regulation. Any drug that pushes that system too hard in one direction should be assumed risky until proven otherwise.
Systems biology supports this caution. A 2025/2026 integrative network analysis identified CB1 and CB2 as highly influential nodes in the endocannabinoid system and connected receptor signaling to broader metabolic pathways. That finding argues against receptor-centric simplification. Altering CB1 does not just shift one synapse. It can propagate across endocrine, immune, metabolic, and behavioral networks.
Lessons from centrally active versus peripherally restricted strategies
The clearest lesson from the first era of CB1 drug development is that central exposure is often where efficacy and liability become inseparable. Centrally active CB1 agonists can engage pain and appetite circuits, but the same brain penetration raises the probability of intoxication-like effects, sedation, cognitive disruption, and psychiatric adverse events. Centrally active CB1 antagonists illustrate the opposite failure mode: they can hit metabolic goals while producing unacceptable mood and anxiety effects because they also block endogenous cannabinoid tone in limbic circuits.
That history is why peripherally restricted CB1 ligands became attractive. The idea is straightforward: if some therapeutic benefits depend on CB1 in peripheral tissues such as primary afferents, gut, liver, adipose tissue, or other non-CNS sites, then keeping a drug out of the brain might preserve useful effects while lowering central adverse effects. For pain, this could mean acting on peripheral nociceptive mechanisms. For metabolic disease, it could mean reducing lipogenesis or altering glucose-related pathways without disturbing mood or cognition.
The logic is sound, but it is not a magic solution. First, peripheral and central CB1 biology are not cleanly separable in disease states. Chronic pain, feeding, and stress all involve brain-body loops. Second, blood-brain barrier exclusion is a quantitative problem, not a binary one. Small amounts of CNS penetration may still matter, especially with chronic dosing. Third, even a truly peripheral CB1 ligand may fail clinically if the targeted disease depends heavily on central circuits or if compensatory pathways blunt efficacy.
The newer strategy is not merely “stay out of the brain.” It is “shape the signal.” That includes partial agonists, neutral antagonists rather than inverse agonists, allosteric modulators, and biased ligands that favor selected downstream pathways over others. The 2025 American Journal of Psychiatry discussion of CB1 biased signaling in schizophrenia fits here. If a ligand can preserve selected therapeutic signaling while reducing beta-arrestin recruitment, receptor desensitization, or unwanted circuit effects, then CB1 might become more tractable. Might. There is still no guarantee that pathway bias observed in cell systems will map neatly onto human therapeutic windows.
So the current view of CB1 as a drug target is neither dismissive nor romantic. CB1 is appealing because it regulates synaptic gain at precisely the kinds of circuits clinicians want to influence. It is difficult because those circuits also govern memory, affect, motivation, and stress resilience. Peripherally restricted ligands, selective signaling strategies, and better structural understanding have improved the design logic. They have not erased the core problem. A receptor this central to brain function rarely tolerates blunt pharmacology.
CB2 as a drug target: neuroinflammation, neurodegeneration, and immune modulation
CB2 has become one of the most tempting targets in cannabinoid pharmacology for a simple reason: it appears to offer access to immune and glial biology with less risk of the intoxication, memory impairment, and abuse liability classically tied to strong CB1 activation. That appeal matters well beyond academic receptor mapping. The World Health Organization estimated that 200 million people used cannabis in 2019, or 4% of the global population aged 15–64, and the U.S. FDA still lists only one cannabis-derived drug product and three cannabis-related drug products as approved as of 2025. Interest is high. Translation is hard.
The older shorthand said CB1 is the brain receptor and CB2 is the immune receptor. That is now too crude to be useful. The 2026 Frontiers in Behavioral Neuroscience review, explicitly framed as “an update over the last 3 years,” treats CB2 as relevant to central nervous system disorders precisely because it participates in neuroinflammatory and neurodegenerative mechanisms, including microglial activation, cytokine signaling, and neuron-glia crosstalk. The key shift is not that CB2 suddenly became abundant everywhere in the brain. It did not. The shift is that low basal expression in many CNS regions does not mean functional irrelevance, especially when disease states can change receptor expression, trafficking, and signaling.
Why CB2 is often viewed as a safer target
The safety argument for CB2 starts with distribution, but it does not end there. CB2 is enriched in immune cells and is often upregulated in activated microglia, infiltrating macrophages, and other inflammatory cell populations. By contrast, CB1 is densely expressed at presynaptic terminals across cortex, hippocampus, basal ganglia, and cerebellum, where it strongly shapes neurotransmitter release. That anatomical difference matters because a ligand that mainly engages CB2 is less likely to reproduce the classic psychoactive profile associated with CB1.
Less likely is not the same as impossible. Selectivity is a moving target. The 2026 Frontiers in Chemical Biology review stresses that “CB1 and CB2” selectivity depends on structural differences that alter binding, efficacy, and receptor regulation, and a recent PubMed-indexed study on subtype selectivity argues that endocannabinoid discrimination between receptor subtypes reflects conformational dynamics rather than a fixed lock-and-key model. In practice, that means a compound can look CB2-selective in one assay and behave less cleanly in another, especially when receptor density, membrane environment, and downstream transducers differ. The same ligand may bias G-protein signaling in one cell type, recruit beta-arrestins in another, and show different desensitization or internalization kinetics under inflammatory conditions.
That mechanistic complexity actually strengthens the case for pursuing CB2 carefully. If the therapeutic goal is to reduce microglial cytokine output, limit peripheral immune cell recruitment, or alter glial metabolic stress without strongly perturbing cortical synaptic transmission, then CB2 remains the more plausible first receptor to target. CB1 biased signaling, as argued in the 2025 American Journal of Psychiatry article on schizophrenia, may eventually widen the therapeutic window for CB1-directed drugs. But that is an argument for smarter CB1 pharmacology, not a reason to ignore the lower psychoactivity liability of CB2-focused approaches.
Safer, though, should not be confused with effective. A drug can avoid obvious intoxication and still fail because the receptor is expressed too sparsely, too variably, or too transiently in the disease process to generate a meaningful clinical effect. That has happened repeatedly across neuroinflammation drug development, not just in cannabinoid science.
Evidence and limits in CNS disease models
The Frontiers in Behavioral Neuroscience review lays out why CB2 keeps returning in models of Alzheimer’s disease, Parkinson’s disease, multiple sclerosis, traumatic brain injury, stroke, and neuropathic pain: these conditions all include inflammatory signaling, glial reactivity, oxidative stress, and progressive tissue damage. In many rodent studies, CB2 agonists reduce pro-inflammatory mediators such as TNF-α, IL-1β, and IL-6, shift microglia toward less damaging phenotypes, or dampen leukocyte infiltration. Sometimes that is accompanied by improved motor behavior, reduced lesion size, or preservation of synaptic markers. Those are real signals, not artifacts to dismiss.
But the record is uneven. Effects often depend on timing. A CB2 agonist given during the early inflammatory wave after injury may do something very different from the same agonist given during chronic degeneration. Dose also matters, as does ligand class. Endocannabinoids such as 2-AG and anandamide are not interchangeable with phytocannabinoids or with synthetic CB2-preferring agonists. Their local concentrations, metabolic breakdown, and off-target actions differ. So do their pathway preferences.
The disease models themselves are another limit. A transgenic Alzheimer’s mouse with amyloid pathology and reactive microglia is not Alzheimer’s disease in a 74-year-old human with mixed vascular, inflammatory, and proteostatic failure. Experimental autoimmune encephalomyelitis is useful for multiple sclerosis biology, but it captures only part of the human condition. Positive preclinical results therefore establish plausibility, not proof.
There is also a persistent measurement problem in CB2 biology. Antibody specificity has long been a weak point, making receptor localization claims harder to trust than many papers imply. Low expression levels in the healthy brain, inducible expression in disease, and species differences all complicate interpretation. Some reported “neuronal CB2” findings may be real in specific contexts, but broad claims of widespread constitutive neuronal CB2 expression remain less secure than the field once suggested.
This is where signaling matters as much as anatomy. CB2 is a Gi/o-coupled GPCR, but that shorthand hides important variance: inhibition of adenylyl cyclase, modulation of MAPK pathways, effects on ion channels, beta-arrestin recruitment, receptor desensitization, and internalization can all change the net biological outcome. A ligand that reduces cytokine release in cultured microglia may fail in vivo if it rapidly desensitizes receptors or if it triggers an unhelpful arrestin-dominant program. Lower liability for intoxication does not solve these pharmacology problems.
What the recent literature suggests for future therapies
The recent literature points away from the old search for a single “CB2 agonist for brain inflammation” and toward a more selective, context-dependent strategy. The 2026 structural review and the 2025/2026 subtype-selectivity paper both support the same lesson: receptor targeting will depend on conformational state, efficacy profile, and tissue context, not just nominal subtype affinity. Drug design is moving toward ligands that are selective in function, not merely in binding.
That likely means three things. First, future CB2 drugs will need better signaling fingerprints, including G-protein versus beta-arrestin bias, receptor residence time, and cell-type-specific effects in human systems rather than only immortalized assay lines. Second, disease stratification will matter. A therapy aimed at CB2 may work best where inflammatory amplification is a major driver, not just a byproduct, of pathology. Third, combination thinking is unavoidable. Integrative network analysis published in a recent PubMed-indexed study identified CB1 and CB2 as influential nodes across broader endocannabinoid and metabolic pathways, which fits the lived reality of CNS disease: inflammation, mitochondrial stress, lipid signaling, and synaptic dysfunction are entangled.
Under specific inflammatory conditions, CB2 modulation may reduce damaging immune signaling while avoiding much of the CB1-linked psychoactive burden.Preliminary evidence
That does not make CB2 a dead end. It makes it a target that probably belongs in precision pharmacology rather than receptor mythology. The strongest current case is not “CB2 activation treats neurodegeneration.” It is narrower: under specific inflammatory conditions, in specific cell populations, with the right ligand properties, CB2 modulation may reduce damaging immune signaling while avoiding much of the CB1-linked psychoactive burden. That is a credible therapeutic hypothesis. It is not yet a clinically validated rule.
Cannabis compounds at CB1 and CB2: THC, CBD, minor cannabinoids, and synthetic ligands
Cannabis does not act through a single chemical or a single receptor mechanism. That point sounds obvious, yet public discussion still collapses the pharmacology into a false pairing: THC equals CB1, CBD equals CB2. The literature does not support that shortcut. Cannabis contains many phytocannabinoids, plus terpenes, flavonoids, and other constituents, while the body makes its own endocannabinoids such as anandamide and 2-arachidonoylglycerol, identified through work associated with Raphael Mechoulam, Lumír Hanuš, and others. On top of that, most of what scientists know about receptor activation, efficacy, selectivity, desensitization, and signaling bias comes from synthetic ligands designed as probes or drug leads rather than from plant compounds alone.
That distinction matters because CB1 and CB2 are not passive locks waiting for a cannabinoid key. They are class A GPCRs with multiple conformational states, coupling preferences, and regulatory fates. A 2026 structural review in Frontiers in Chemical Biology makes the point clearly: ligand selectivity at CB1 and CB2 depends on receptor-level structural differences that change binding, efficacy, and receptor regulation, not just whether a molecule “hits” one receptor or the other. A recent PubMed-indexed study on subtype selectivity likewise argues that endocannabinoid preference for CB1 versus CB2 arises from conformational dynamics rather than simple lock-and-key fit. So when two cannabinoids look chemically similar but produce different effects in brain, immune tissue, or pain circuits, that is not a side note. It is the core pharmacology.
Given that the World Health Organization estimated 200 million people used cannabis in 2019, about 4% of the global population aged 15 to 64, these distinctions are not academic trivia. They shape intoxication, therapeutic claims, safety, and why some receptor-targeted drugs succeed while others fail.
How THC engages CB1 and contributes to psychoactive effects
Delta-9-tetrahydrocannabinol, or THC, is still the central compound for understanding cannabis intoxication because it directly engages CB1 in the central nervous system. That is the cleanest receptor-level explanation for the characteristic psychoactive effects of cannabis, even though “psychoactive” covers several separable outcomes: altered time perception, reward, memory disruption, anxiogenesis in some users, and impaired motor coordination. Allyn Howlett’s receptor pharmacology work helped establish this CB1-centered framework, and it remains foundational.
But “THC activates CB1” is only the start. THC is not a perfect on-switch. It is generally described as a partial agonist at CB1, meaning its efficacy depends on receptor density, tissue context, and the signaling assay used. In high-CB1 regions such as cortex, hippocampus, basal ganglia, and cerebellum, that can be enough to produce strong central effects. At the synaptic level, CB1 is often presynaptic, where activation suppresses neurotransmitter release. That means THC can dampen glutamate release in one circuit, GABA release in another, and produce opposite network-level outcomes depending on which cells are expressing the receptor and when.
This is why receptor location cannot be reduced to “CB1 in the brain.” CB1 is abundant in brain, yes, but not evenly distributed, not static, and not functionally identical across neuron types. The result is that THC’s effects are circuit-dependent. Euphoria and reinforcement are part of the picture. So are short-term memory disruption and psychosis-relevant signaling in susceptible people.
That last point has become more important, not less. A 2025 American Journal of Psychiatry article argued that CB1 biased signaling is a plausible therapeutic strategy in schizophrenia, precisely because CB1 biology cannot be understood as one-dimensional receptor activation. Schizophrenia affects about 24 million people worldwide according to WHO, and any serious discussion of THC at CB1 has to account for the fact that the same receptor family implicated in intoxication is also being studied through biased agonism, beta-arrestin recruitment, and pathway-selective drug design. THC may engage Gi/o-coupled signaling, but CB1 also undergoes phosphorylation, desensitization, and internalization, with consequences for tolerance and downstream response. Dose and timing matter. Repeated exposure changes the system.
Synthetic CB1 ligands made this plain long ago. Compounds such as CP55,940 and WIN55,212-2 are potent research agonists that often produce stronger or cleaner receptor responses than THC in laboratory assays. Rimonabant, a CB1 inverse agonist, showed the opposite direction: blocking or pushing CB1 below basal activity could reduce appetite and body weight, but psychiatric adverse effects sank the drug. That clinical story is a warning. Selective CB1 targeting can be pharmacologically elegant and still medically disappointing.
Why CBD does not fit a simple CB1/CB2 agonist story
CBD is the cannabinoid most often flattened into myth. It is routinely presented as “the non-intoxicating one that works through CB2” or as THC’s direct receptor opposite. Neither claim is accurate.
| Compound class or example | How the article describes receptor behavior | Key takeaway |
|---|---|---|
| THC | Partial agonist at CB1 and CB2; central to intoxication through CB1 | CB1 engagement helps explain psychoactive effects, but outcome is still context-dependent |
| CBD | Not a straightforward CB1 or CB2 agonist; may act allosterically or indirectly | CBD is not a simple CB2 mirror image of THC |
| Minor cannabinoids | Reported effects vary by assay and concentration | Do not generalize receptor behavior from one cannabinoid to all |
| Synthetic ligands | Often used to define efficacy, selectivity, and bias | Probe compounds can behave very differently from plant cannabinoids |
CBD does not behave like a straightforward CB1 or CB2 agonist in the way THC does at CB1. Its affinity at the orthosteric sites of CB1 and CB2 is relatively low, and much of its pharmacology appears to involve indirect, allosteric, or non-cannabinoid-receptor actions. Depending on the system studied, CBD has been reported to act as a negative allosteric modulator at CB1, which could alter how THC or endocannabinoids signal without simply turning the receptor on or off. That distinction is crucial. Modulating receptor shape and signaling efficiency is not the same thing as classical agonism.
CBD also interacts with targets beyond CB1 and CB2, including TRPV1, 5-HT1A, PPAR-gamma, adenosine-related pathways, and enzymes involved in endocannabinoid tone. Some effects may reflect altered anandamide signaling rather than direct receptor stimulation. That helps explain why CBD’s clinical profile does not line up neatly with the old receptor shorthand. The U.S. FDA notes that it has approved one cannabis-derived drug product and three cannabis-related drug products as of 2025; CBD’s best-established medical use sits in epilepsy, a condition affecting around 50 million people worldwide, and that therapeutic path cannot be explained by a cartoon model of “CBD binds CB2 and reduces inflammation.”
Even for CB2, the picture is broader than the older peripheral narrative. A 2026 Frontiers in Behavioral Neuroscience review described “an update over the last 3 years” in which CB2 signaling gained attention in central nervous system disorders through links to neuroinflammatory and neurodegenerative mechanisms. That does not mean CBD is simply a CB2 drug. It means the receptor itself has a larger functional footprint than the public story suggests.
The importance of separating plant compounds from receptor myths
Phytocannabinoids are not interchangeable, and they are not interchangeable with synthetic ligands. Minor cannabinoids such as cannabigerol (CBG), cannabinol (CBN), tetrahydrocannabivarin (THCV), and cannabichromene (CBC) have been reported to interact with CB1 and CB2 in ways that vary by assay and concentration. Some show weak agonism, some partial agonism, some antagonism under certain conditions, and some meaningful activity at non-cannabinoid targets. THCV is a good example: it has been described as a CB1 antagonist or neutral antagonist at low doses in some systems and a partial agonist at higher doses in others. That is not inconsistency for its own sake. It reflects context-dependent receptor pharmacology.
Research tools sharpen this point. Endocannabinoids, phytocannabinoids, and synthetic ligands can all activate “CB1 and CB2,” but they do not stabilize the same receptor conformations or produce the same bias across G proteins and beta-arrestins. The 2026 structural review and recent network analyses indexed in PubMed both push the field away from receptor mythology and toward systems pharmacology. Receptors sit inside cell-specific signaling networks, metabolic pathways, and trafficking machinery. A ligand’s clinical profile emerges from that full setting, not from a label like “CB2 anti-inflammatory” or “CBD balances THC.”
For a cannabis wiki, that is the line that matters most: THC has primary relevance to CB1-mediated intoxication, but cannabis pharmacology does not end there, and CBD is not a simple CB2 mirror image of THC. Plant compounds start the story. Receptor state, tissue distribution, efficacy, bias, and regulation decide how it ends.
Schizophrenia, psychosis risk, and what CB1 biology does and does not imply
Why schizophrenia enters the receptor conversation
Schizophrenia matters here because CB1 is not just another receptor on a long target list. It is one of the most abundant G protein-coupled receptors in the brain, positioned on presynaptic terminals where it can dampen transmitter release and reshape network activity in circuits already implicated in psychosis: cortex, hippocampus, striatum, amygdala. When a receptor sits that close to glutamate, GABA, dopamine-related circuit control, and stress responsivity, psychiatric researchers will pay attention.
The public-health scale makes this more than an academic question. The World Health Organization estimates that cannabis was used by 200 million people in 2019, or 4% of the global population aged 15–64. WHO also estimates that schizophrenia affects about 24 million people worldwide, roughly 1 in 300 people. Those are not small, isolated populations. They overlap in real clinics, in emergency departments, and in longitudinal cohort studies. Any receptor biology that might illuminate psychosis risk, antipsychotic drug development, or both deserves serious scrutiny.
That is the translational backdrop for the 2025 American Journal of Psychiatry paper arguing that CB1 biased signaling is a plausible therapeutic strategy for schizophrenia. The core idea is easy to state and easy to misuse. Easy to state: CB1 does not behave like a simple on/off switch, and different ligands can stabilize different receptor conformations, shifting signaling toward or away from specific intracellular pathways. Easy to misuse: if CB1 is relevant to schizophrenia treatment research, some readers jump to the false claim that “cannabinoid activation helps schizophrenia” or that cannabis use is therefore medicinal by default. The paper does not support that leap.
Historically, cannabinoid science got stuck in shorthand. CB1 was framed as the “brain receptor,” CB2 as the “immune receptor,” and psychoactive effects were pinned almost entirely on CB1 occupancy. That framing was always incomplete, and by 2025 it is plainly too crude. Foundational work by Allyn Howlett established cannabinoid receptor pharmacology, while Mechoulam and Hanus helped define endocannabinoid biology through anandamide. Since then, receptor pharmacology has moved from receptor presence to receptor state. That shift matters for schizophrenia because pathophysiology is not caused by receptor expression alone. It emerges from timing, cell type, circuit context, and intracellular response.
The newer literature strengthens that point. A 2026 Frontiers in Chemical Biology review explains that structural differences between CB1 and CB2 shape ligand binding, efficacy, signaling, and receptor regulation. A 2025/2026 PubMed-indexed study on subtype selectivity argues that endocannabinoid selectivity can arise from conformational dynamics rather than a static lock-and-key model. Another 2025/2026 network-analysis paper identifies CB1 and CB2 as highly influential nodes across the endocannabinoid system, linked to metabolic and signaling pathways rather than isolated receptor boxes. For schizophrenia research, this means one should stop asking only “does a compound hit CB1?” and start asking “which CB1 states, in which cells, under which exposure conditions?”
That is where the clinical seriousness comes from. Not slogan-level receptor talk. Mechanistic precision.
Biased signaling versus blunt receptor activation
Blunt CB1 activation is a poor model for therapeutic reasoning. It collapses too many variables into one word: activation. Endogenous ligands such as anandamide and 2-AG, phytocannabinoids such as delta-9-THC, and synthetic agonists do not behave identically at CB1, even when they all count as ligands. They can differ in affinity, efficacy, residence time, regional exposure, and pathway bias. They can also differ in how strongly they recruit Gi/o proteins versus beta-arrestins, how quickly they trigger desensitization, and how much receptor internalization follows repeated dosing.
That distinction is central to the AJP schizophrenia argument. A ligand that nudges CB1 toward one intracellular program may not resemble a ligand that drives broad receptor activation across many brain regions. In GPCR pharmacology, that difference can separate a useful drug from an intolerable one. CB1 is especially sensitive to this problem because the same receptor can reduce excitatory transmission in one synapse, suppress inhibitory transmission in another, and alter downstream plasticity in ways that are not functionally equivalent. Small molecular differences can produce large systems-level consequences.
THC is the obvious example of why “CB1 target” does not equal “CB1 therapy.” THC is a phytocannabinoid with partial agonist properties at CB1, but the lived and clinical effects of THC are not a pure readout of one signaling branch. Dose matters. Age at exposure matters. Repeated exposure matters. So does the ratio of THC to other constituents, and whether the person has psychiatric vulnerability. Acute intoxication-like effects, anxiety, perceptual change, and transient psychotic symptoms in some users do not tell us that all CB1-directed therapeutics are doomed. They do tell us that indiscriminate CB1 stimulation is not a shortcut to antipsychotic treatment.
Biased agonism is attractive precisely because it tries to avoid that bluntness. The therapeutic hope is not “more CB1.” It is “the right CB1 signaling profile.” That may mean favoring signaling routes linked to circuit stabilization while limiting routes linked to adverse cognitive or psychotomimetic effects, tolerance, or receptor downregulation. Whether this can be achieved in patients is still an open question. The concept is plausible. It is not proven clinical success.
CB2 belongs in this picture too, though not as a simplistic fix. The 2026 Frontiers in Behavioral Neuroscience review describes “an update over the last 3 years” in which CB2 signaling gained attention in central nervous system disorders because of neuroinflammatory and neurodegenerative links. That weakens the old claim that CB2 is irrelevant to brain disease. But it does not rescue the old binary by flipping it. Schizophrenia is not just inflammation, and CB2 is not just an anti-inflammatory button. Any serious translational model has to account for neuron-glia interactions, immune signaling, and circuit dysfunction together.
What not to infer
- Not a proof of benefit CB1 being a drug target does not mean cannabis use protects against schizophrenia or psychosis.
- Not a rebuttal of risk Mechanistic receptor studies do not cancel epidemiologic concern about high-THC exposure in vulnerable people.
- Not interchangeability Endocannabinoids, phytocannabinoids, and synthetic ligands should not be treated as the same kind of exposure.
- Not blanket approval Existing FDA approvals do not settle psychiatric safety or efficacy questions for broad cannabinoid use.
What readers should not infer from mechanistic studies
First, readers should not infer that because CB1 is a therapeutic target, cannabis use is therefore protective against schizophrenia or psychosis. Drug-target logic does not work that way. Beta receptors are drug targets; that does not mean any stimulant is a treatment. Opioid receptors are drug targets; that does not mean all opioid agonism is beneficial. The same principle applies here.
Second, mechanistic studies do not erase epidemiologic concern. Research on receptor bias and ligand design sits alongside, not against, evidence that cannabis exposure can worsen psychosis risk in some people, especially with high-THC products, early initiation, or heavy use. A receptor mechanism may explain why some compounds could be engineered for better outcomes. It does not justify broad public claims that “CB1 activation is good for schizophrenia.”
Third, readers should not treat endocannabinoids, phytocannabinoids, and synthetic ligands as interchangeable. Anandamide is not THC. A peripherally restricted CB1 modulator is not smoked cannabis. A low-efficacy, pathway-biased investigational compound is not a high-exposure consumer product. These categories matter because receptor behavior depends on ligand class, dose, route, timing, and tissue exposure.
Finally, the existence of approved cannabis-related medicines does not settle psychiatric questions. As of 2025, the FDA notes one cannabis-derived drug product and three cannabis-related drug products have been approved. That fact shows cannabinoid pharmacology can become medicine under defined conditions. It does not mean unrefined receptor stimulation is safe, effective, or appropriate for schizophrenia.
The correct takeaway is stricter and less comforting. CB1 biology is relevant to psychosis because the receptor is deeply embedded in brain circuits that shape salience, cognition, and inhibition. That relevance supports careful drug design, not casual extrapolation. In schizophrenia research, receptor pharmacology is a map of possibilities and liabilities at the same time.
What receptor science means for future cannabinoid medicines
Cannabinoid medicine is moving away from the old idea that a useful drug merely needs to “hit CB1” or “hit CB2.” That shorthand was always incomplete. It is now actively misleading. CB1 and CB2 are not simple on-off switches split neatly into brain versus immune system. They are receptors with overlapping but uneven distributions across cell types, dynamic conformations, distinct coupling preferences, and changing roles across disease states. That matters because the same ligand can look therapeutic in one tissue, intoxicating in another, and ineffective in a clinical trial if the disease population is too broad or the dosing schedule drives desensitization.
The public-health stakes are not minor. The World Health Organization estimated that 200 million people used cannabis in 2019, equal to 4% of the global population aged 15 to 64. Yet the U.S. FDA still notes only one cannabis-derived drug product and three cannabis-related drug products have been approved as of 2025. That gap between widespread exposure and limited approved therapeutics is one reason receptor pharmacology matters so much. Another is disease burden: epilepsy affects about 50 million people worldwide, schizophrenia about 24 million, and chronic pain affects nearly 1 in 5 adults in the United States. If cannabinoid biology is going to yield better medicines, it will not happen through crude receptor activation.
Foundational work by Raphael Mechoulam, Lumír Hanuš, and Allyn Howlett established the modern field by identifying endocannabinoid signaling and receptor pharmacology. The next phase is different. It is less about proving CB1 and CB2 exist, and more about learning how to steer them precisely.
| Design concept | What it aims to improve | Why the article says it matters |
|---|---|---|
| Subtype selectivity | Prefer CB1 or CB2 more cleanly | Subtype alone does not guarantee efficacy or safety |
| Efficacy tuning | Avoid overly strong receptor activation | Different active states can change benefit, tolerance, and adverse effects |
| Biased signaling | Favor one intracellular pathway over another | May help separate useful effects from liabilities |
| Allosteric modulation | Tune receptor response to endogenous ligands | Could preserve more normal spatial and temporal signaling |
| Tissue restriction | Limit exposure to selected organs or avoid brain entry | Useful when central adverse effects dominate |
Selectivity, efficacy, allosteric modulation, and bias
The most important lesson from recent receptor science is that “binding” is not the same as “effect.” A ligand can prefer CB1 over CB2, or vice versa, but even that is only the start. It can also differ in efficacy, meaning how strongly it stabilizes active receptor states. It can favor Gi/o signaling over beta-arrestin recruitment, or promote receptor internalization more than sustained surface signaling. Those differences can change outcomes dramatically.
The 2026 Frontiers in Chemical Biology review on cannabinoid receptor structure makes this point clearly: subtype selectivity between CB1 and CB2 depends on receptor-level structural differences that shape ligand binding, signaling efficacy, and receptor regulation. That is why chemically similar cannabinoids do not behave identically. A phytocannabinoid, an endocannabinoid like anandamide, and a synthetic ligand may all contact the same receptor family while producing different downstream profiles. Drug design that ignores efficacy and signaling bias is now behind the evidence.
CB1 is the clearest case. It is often discussed as the receptor responsible for psychoactive effects, which is true but incomplete. CB1 also regulates synaptic transmission, pain processing, appetite, stress circuits, and cortical signaling. The American Journal of Psychiatry article from 2025 argues that biased CB1 signaling is a plausible therapeutic strategy for schizophrenia. That is a striking shift in the field. Schizophrenia affects roughly 24 million people worldwide, and the article does not treat CB1 as a receptor to avoid altogether. Instead, it treats CB1 as a target whose pathological and therapeutic consequences may depend on which intracellular pathways a ligand favors. That is classic GPCR pharmacology, now applied seriously to cannabinoid therapeutics.
CB2 has undergone a similar reappraisal. The 2026 Frontiers in Behavioral Neuroscience review describes “an update over the last 3 years” showing growing interest in CB2 signaling in central nervous system disorders, especially where neuroinflammatory and neurodegenerative mechanisms are involved. That does not mean CB2 is a magic anti-inflammatory target. It means the old claim that CB2 is irrelevant to the brain no longer fits the literature. Expression can be low under baseline conditions and increase in particular cell populations or disease contexts. A CB2-directed drug may therefore depend heavily on timing, pathology, and tissue state.
Allosteric modulation may become one of the most useful ways to exploit these distinctions. Orthosteric agonists often drive broad receptor activation, which raises the risk of side effects, tolerance, and poor context selectivity. Allosteric modulators, by contrast, can tune how receptors respond to endogenous ligands rather than forcing maximal activation. In principle, that could preserve spatial and temporal aspects of normal endocannabinoid signaling. It is an appealing strategy for CB1, where direct agonism often runs into central adverse effects, and for CB2, where disease-linked upregulation may make context-dependent modulation more attractive than constant stimulation. But “appealing” is not “proven.” Many allosteric ideas that look elegant in vitro still fail once receptor reserve, tissue heterogeneity, and pharmacokinetics enter the picture.
Why structural and network biology now guide drug design
The field’s center of gravity has shifted from simple receptor mapping to dynamic receptor-state analysis. This is where structural biology matters. The 2026 Frontiers review and the recent PubMed-indexed study on the dynamic mechanism of subtype selectivity both push against a lock-and-key model. Selectivity is not just a matter of one ligand fitting one receptor pocket better. Receptors breathe. Ligands stabilize ensembles of conformations. Endocannabinoids can achieve subtype preference through dynamic interactions that alter how CB1 or CB2 moves between inactive, active, and signaling-competent states.
That change in thinking has practical consequences. A medicinal chemist no longer asks only, “Does this compound bind CB1 or CB2?” The better questions are: Which conformations does it stabilize? Does it promote Gi/o more than beta-arrestin? Does it trigger rapid internalization? Does it behave differently in neurons, microglia, hepatocytes, or peripheral nociceptors? Can it be restricted to the periphery? Does it cooperate with endogenous ligand tone that rises only during inflammation or injury?
Network biology extends this even further. A recent integrative network analysis indexed in PubMed identified CB1 and CB2 as highly influential nodes within the endocannabinoid system and linked receptor signaling to broader metabolic pathways. That matters because endocannabinoid pharmacology is never only about receptors. Anandamide and 2-AG synthesis, transport, and degradation shape receptor exposure. So do lipid metabolism, inflammatory mediators, and cross-talk with other GPCR and ion-channel systems. A ligand that looks clean in a recombinant assay can behave very differently in living tissue where FAAH, MAGL, prostaglandin pathways, and local receptor density all vary.
This systems view also helps explain why clinical translation has been uneven. Some failures likely came from bad compounds. Others came from oversimplified disease models. If a trial enrolls patients whose symptoms share a label but not an endocannabinoid signature, the average result may look disappointing even if a biologically defined subgroup could benefit. Future success will depend not just on chemistry, but on stratification: which patients, which disease stage, which tissues, which receptor state, which endogenous tone.
Realistic expectations for the next generation of therapies
The next generation of cannabinoid medicines is likely to be narrower, more selective, and less romantic than public discussion suggests. That is good news. Better drugs often emerge when a field gives up on one-size-fits-all explanations.
Some progress is already visible. Peripheral CB1 restriction remains attractive for pain or metabolic indications because it aims to reduce central adverse effects. CB2-directed compounds remain plausible for inflammatory, neuropathic, and neuroimmune conditions, especially if disease-linked expression creates therapeutic windows. Biased CB1 ligands may prove useful in neuropsychiatric disease if they can separate desired circuit effects from unwanted intoxication, anxiety, or tolerance. Allosteric modulators may offer subtler control than direct agonists. Enzyme-targeting approaches that alter endocannabinoid tone indirectly may still be valuable, though they carry their own risks and do not bypass receptor complexity.
Still, realism is essential. Selectivity does not guarantee success. A highly selective CB2 agonist can still fail if CB2 expression in the target disease is too variable or if downstream signaling is not the real driver of symptoms. Bias measured in one assay may not predict bias in human tissue. Structural snapshots are powerful, but they are snapshots. Tissue restriction can reduce some liabilities while creating others. And phytocannabinoids should not be treated as stand-ins for all cannabinoid pharmacology; endocannabinoids, plant-derived compounds, and synthetic ligands can produce very different receptor behaviors depending on dose, timing, and signaling context.
So the sober forecast is this: future cannabinoid medicine will probably improve by becoming more precise, not more broadly activating. The most credible paths forward combine receptor selectivity, signaling bias, allosteric control, tissue targeting, and better patient stratification. What is known is substantial: CB1 and CB2 are dynamic signaling hubs, structural state matters, and disease context matters. What remains uncertain is whether that mechanistic clarity will translate into repeatable clinical gains across pain, psychiatric illness, epilepsy, neurodegeneration, and inflammatory disease. The field is still medically important because the unmet need is large, the biology is real, and the older simplistic model has finally given way to a pharmacology capable of explaining both promise and disappointment.
References
- [1]Cannabis (marijuana). WHO Fact Sheet, 2024. https://www.who.int/news-room/fact-sheets/detail/cannabis-(marijuana)
- [2]Epilepsy. WHO Fact Sheet, 2024. https://www.who.int/news-room/fact-sheets/detail/epilepsy
- [3]Schizophrenia. WHO Fact Sheet, 2022. https://www.who.int/news-room/fact-sheets/detail/schizophrenia
- [4]FDA Regulation of Cannabis and Cannabis-Derived Products, Including Cannabidiol (CBD). FDA Public Health Focus, 2025. https://www.fda.gov/news-events/public-health-focus/fda-regulation-cannabis-and-cannabis-derived-products-including-cannabidiol-cbd
- [5]The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research. National Academies Press, 2017. https://nap.nationalacademies.org/catalog/24625/the-health-effects-of-cannabis-and-cannabinoids-the-current-state