






Warum CB1 und CB2 in der cannabis-Forschung wichtig sind
GPCR G-Protein-gekoppelter Rezeptor: ein Membranrezeptor, der nach Ligandenbindung seine Form verändert und über intrazelluläre Partner wie G-Proteine und Beta-Arrestine Signale weiterleitet.
Was die Rezeptorantwort bestimmt
- Ort Gewebe, Zelltyp und subzelluläre Position prägen die Antwort.
- Ligandenidentität Endocannabinoide, Phytocannabinoide und synthetische Liganden lösen keine identischen Signalzustände aus.
- Verfügbare Partner Verschiedene Zellen bieten unterschiedliche G-Proteine, Kinasen und Beta-Arrestine.
- Expositionsmuster Signaldauer und wiederholte Stimulation beeinflussen Desensibilisierung und Internalisierung.
- Signalweg-Bias Ein Ligand kann G-Protein-Signalisierung, Beta-Arrestin-Rekrutierung oder andere Ausgänge bevorzugen.
Die öffentliche Kurzformel hat die Biologie der cannabinoid-Signale lange auf eine einfache Zweiteilung reduziert: CB1 erklärt den „High“, CB2 kümmert sich irgendwo außerhalb des Gehirns um Entzündungen. Diese Darstellung ist zu grob, um nützlich zu sein. CB1 und CB2 sind G-Protein-gekoppelte Rezeptoren, oder GPCRs, und wie andere GPCRs funktionieren sie nicht als einfache Ein-/Aus-Schalter. Sie übersetzen Signale von Endocannabinoiden, die der Körper selbst bildet, von Phytocannabinoiden aus Cannabis sativa und von synthetischen Liganden, die in Laboren entwickelt wurden, in veränderliche zelluläre Antworten. Welche Antwort auftritt, hängt davon ab, wo der Rezeptor sitzt, welcher Ligand bindet, welche Signalisierungspartner verfügbar sind, wie lange der Rezeptor stimuliert wird und ob der Rezeptor in Richtung G-Protein-Signalisierung, β-Arrestin-Rekrutierung, Desensibilisierung oder Internalisierung gelenkt wird.
Das ist deshalb wichtig, weil es in der cannabis-Wissenschaft nicht nur um Intoxikation geht. Es geht auch um chronische Schmerzen, Epilepsie, Immun-Signalisierung, Neurodegeneration, psychiatrische Risiken und darum, weshalb so viele cannabinoid-Arzneiprogramme präklinisch vielversprechend aussahen und dann beim Menschen scheiterten. Die Bedeutung ist groß. Die World Health Organization schätzte, dass 2019 200 Millionen Menschen cannabis konsumierten, etwa 4 % der Weltbevölkerung im Alter von 15 bis 64 Jahren. Epilepsie betrifft weltweit rund 50 Millionen Menschen. Schizophrenie betrifft etwa 24 Millionen. Und dennoch verweist die U.S. FDA Stand 2025 auf die Zulassung eines cannabis-abgeleiteten Arzneimittelprodukts und dreier cannabis-bezogener Arzneimittelprodukte. Diese Lücke zwischen massiver Exposition und begrenzten zugelassenen Therapeutika ist ein Grund, warum die Rezeptor-Biologie so wichtig ist.
| Merkmal | CB1 | CB2 |
|---|---|---|
| Typische Kurzbezeichnung | „Gehirnrezeptor“ | „Immunrezeptor“ |
| Korrektur des Artikels | Zentrale Anreicherung, aber auch periphere Expression | Immunreich, aber für das Gehirn nicht irrelevant |
| Genannte Beispiel-Funktionen | Wahrnehmung, Gedächtnis, Motorik, Nozizeption | Zytokin-Signalisierung, Zellmigration, neuroinflammatorische Rollen |
| Interpretation | Schaltkreis- und zustandsabhängig | Zelltyp- und krankheitszustandsabhängig |
Warum die Rezeptor-Biologie mehr erklärt als Pflanzenetiketten
Bezeichnungen wie „indica“, „sativa“ oder sogar „THC-dominant“ und „CBD-dominant“ erzählen nur einen Teil der Geschichte, weil Rezeptoren und nicht pflanzliche Marketing-Kategorien mechanistisch am nächsten liegen. Δ9-Tetrahydrocannabinol (THC) ist ein partieller Agonist an CB1 und CB2, aber der nachgeschaltete Effekt von THC ist nicht festgelegt. In kortikalen Neuronen mit hoher CB1-Dichte kann es die Neurotransmitter-Freisetzung dämpfen und Wahrnehmung, Gedächtnis und Motorik verändern. In peripheren sensorischen Bahnen kann dieselbe Rezeptor-Familie die Nociception beeinflussen. In Immunzellen kann die Aktivierung von CB2 die Zytokin-Signalisierung oder Zellmigration verschieben. Gleiche Familie. Andere Ergebnisse.
Die einfache Regel, dass CB1 nur im Gehirn und CB2 nur im Immunsystem vorkommt, ist für die heutige Rezeptorbiologie zu grob.Strong evidence
Die alte Faustregel — CB1 im Gehirn, CB2 in Immunzellen — beruhte auf einem realen Muster, ist aber schlecht gealtert. Die Verteilung ist gradientenbasiert und zelltypspezifisch, nicht binär. CB1 wird in vielen Regionen des zentralen Nervensystems stark exprimiert, insbesondere an präsynaptischen Endigungen, kommt aber auch in peripheren Geweben vor. CB2 ist eng mit der Immunfunktion verbunden, doch die Behauptung, es sei für das Gehirn irrelevant, ist nicht mehr haltbar. Eine 2026 in Frontiers in Behavioral Neuroscience veröffentlichte Review argumentierte, dass CB2-Signalisierung bei Störungen des zentralen Nervensystems durch neuroinflammatorische und neurodegenerative Mechanismen zunehmend Beachtung findet, und beschrieb dies als „ein Update der letzten 3 Jahre“. Dieses Update ist bedeutsam. Wenn CB2 unter bestimmten Bedingungen zu zentralen Pathologien beiträgt, dann können auf CB2 zielende Arzneimittel nicht als rein periphere Werkzeuge verstanden werden.
Die Struktur vertieft die Geschichte. Eine 2026 in Frontiers in Chemical Biology veröffentlichte Review erklärte, dass die Liganden-Selektivität zwischen „CB1 und CB2“ aus strukturellen Unterschieden auf Rezeptorebene resultiert, die Bindungslage, Wirksamkeit und Rezeptor-Regulation beeinflussen. In einfachen Worten: Kleine chemische Veränderungen können einen Liganden in Richtung eines Rezeptor-Subtyps oder eines Signalwegs bevorzugen, was erklärt, warum zwei cannabinoid-Verbindungen, die auf dem Papier ähnlich aussehen, in vivo sehr unterschiedlich wirken oder wahrgenommen werden können. Eine 2025/2026 auf PubMed indexierte Studie zur Subtyp-Selektivität ging noch weiter und zeigte, dass die Selektivität von Endocannabinoiden mit der Konformationsdynamik des Rezeptors verknüpft ist und nicht mit einem starren Schlüssel-Schloss-Modell. Der Rezeptor bewegt sich. Der Ligand stabilisiert einige Zustände stärker als andere. Die Biologie folgt diesen Zuständen.
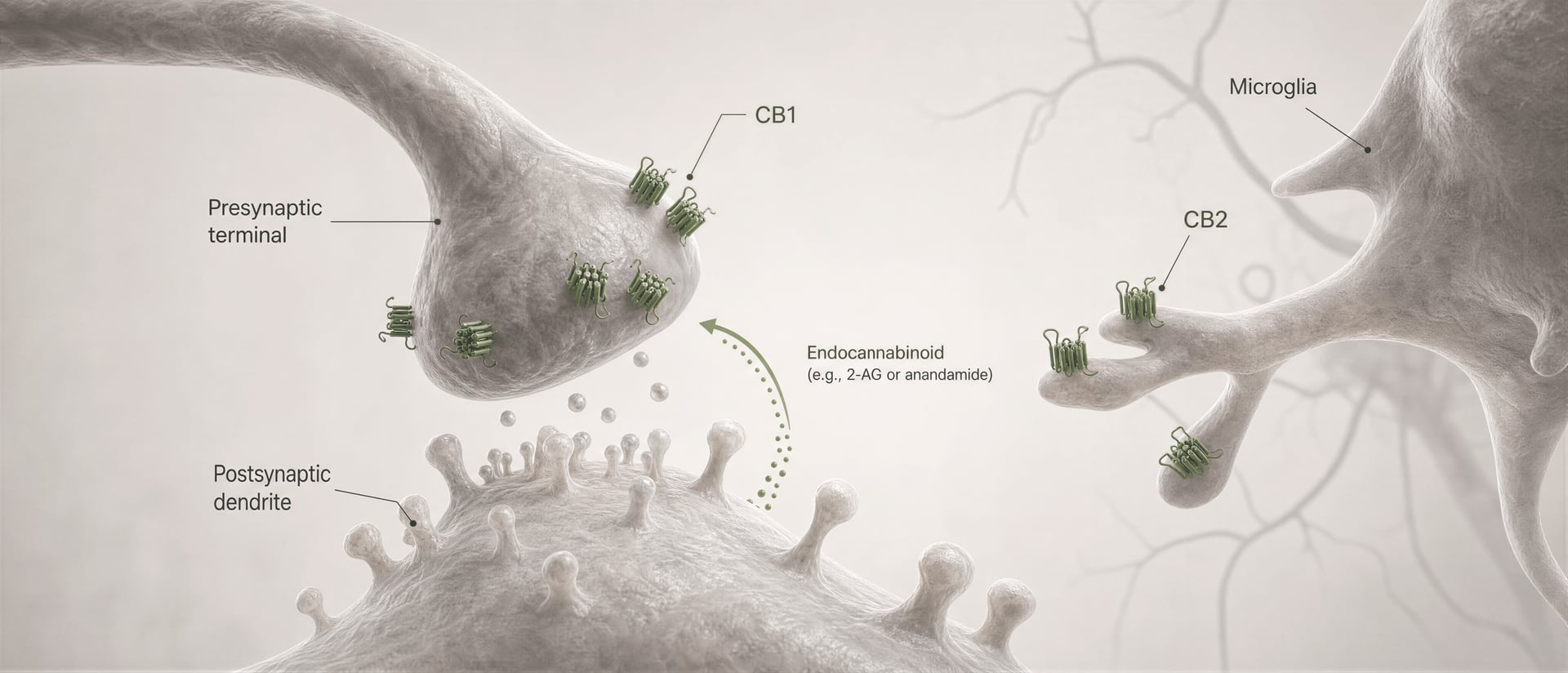
Von Phytocannabinoiden zur Endocannabinoid-Signalisierung
{kind=link}
Retrograde Signalübertragung Ein synaptisches Signalmuster, bei dem eine postsynaptische Zelle einen Botenstoff freisetzt, der rückwärts wirkt und präsynaptische Rezeptoren aktiviert.
Entdeckungsabfolge in Kurzform
- 1 THC wurde isoliert und strukturell definiert.
- 2 Spezifische Cannabinoid-Bindestellen wurden in Hirngewebe nachgewiesen.
- 3 CB1 wurde als GPCR kloniert.
- 4 CB2 wurde aus immunbezogenem Gewebe identifiziert.
- 5 Anandamid und anschließend 2-AG etablierten ein endogenes Signalsystem.
cannabinoid-Rezeptoren wurden nicht entdeckt, weil sich der Körper für cannabis entwickelt hätte. Die Sequenz verlief umgekehrt. Die Arbeit von Allyn Howlett und Kollegen war zentral für die Definition der Pharmakologie von cannabinoid-Rezeptoren, und die spätere Entdeckung von Anandamid durch Raphael Mechoulam und Lumír Hanuš half zu etablieren, dass Menschen ihre eigenen cannabinoid-ähnlichen Signalstoffe produzieren. Anandamid und 2-Arachidonoylglycerol, meist zu 2-AG abgekürzt, sind die wichtigsten Endocannabinoide. Sie werden bei Bedarf gebildet und nicht wie klassische Neurotransmitter in Vesikeln gespeichert, und sie signalisieren oft retrograd: Eine postsynaptische Zelle erzeugt ein Endocannabinoid, das rückwärts über die Synapse wandert, um präsynaptisches CB1 zu aktivieren und die weitere Transmitter-Freisetzung zu verringern.
Das ist ein grundlegend anderes Bild als „Wirkstoff aus Gras trifft Rezeptor“. Endocannabinoid-Signalisierung ist lokal, vorübergehend und eng durch Synthese- und Abbau-Enzyme reguliert. Phytocannabinoide gelangen von außen in dieses System. Synthetische Liganden können es noch stärker oder selektiver ansteuern. Das Ergebnis ist, dass derselbe Rezeptor durch einen flüchtigen endogenen Impuls, durch einen langsam resorbierten oralen Phytocannabinoid oder durch einen synthetischen Agonisten mit hoher Wirksamkeit und sehr unterschiedlichen Sicherheitsrisiken aktiviert werden kann.
Dieser Unterschied ist ein Grund, weshalb Intoxikation nicht allein aus dem Rezeptornamen abgeleitet werden kann. Es hängt von Ligandenwirksamkeit, Dosis, Applikationsweg, Zeitpunkt und Gewebekontext ab. THC an CB1 ist zwar zentral für psychoaktive Effekte, doch dieser Umstand reduziert CB1 nicht auf einen „Psychoaktivitäts-Rezeptor“. Ebenso macht er CB2 nicht zu einem einfachen Entzündungsregler. Der Artikel im American Journal of Psychiatry aus 2025 über CB1-biasierte Signalisierung machte genau diesen umfassenderen Punkt, indem er argumentierte, dass CB1-biasierte Liganden eine therapeutische Strategie für Schizophrenie bieten könnten. Dieser Vorschlag verbindet die cannabis-Wissenschaft mit einer breiteren GPCR-Idee: Wenn ein Ligand günstige Signalzweige bevorzugt, ohne andere zu aktivieren, die mit unerwünschten Wirkungen verbunden sind, könnte sich die Arzneiwirkung von einer groben Rezeptoraktivierung trennen lassen. Ob dieses Versprechen klinisch trägt, bleibt offen, aber die mechanistische Argumentation ist stark.
Was dieser Artikel unter Verteilung, Signalisierung und Arzneizielen versteht
In diesem Artikel bedeutet Verteilung mehr als eine Organübersicht. Sie umfasst Rezeptordichte, Zelltyp, subzelluläre Lokalisation, Krankheitsstatus und zeitliche Veränderung. Ein Rezeptor, der auf GABAergen Endigungen exprimiert wird, kann andere Schaltkreis-Effekte haben als derselbe Rezeptor auf glutamatergen Endigungen. Ein Rezeptor, der während einer Entzündung hochreguliert wird, ist nicht mit seinem Basalzustand gleichzusetzen. Verteilung ist dynamisch.
Signalisierung bedeutet die intrazellulären Folgen der Rezeptorbindung. Für CB1 und CB2 gehören dazu die Kopplung an G-Proteine der Gi/o-Familie, die Hemmung der Adenylylcyclase, die Modulation von Ionenkanälen, Veränderungen von Kinase-Kaskaden, β-Arrestin-Rekrutierung, Rezeptor-Desensibilisierung und Internalisierung. Dazu gehören auch allosterische Modulation und biased agonism, bei denen Liganden bestimmte Signal-Ausgänge gegenüber anderen bevorzugen können. Das ist kein akademisches Detail. Es ist oft der Unterschied zwischen Analgesie, Sedierung, Toleranz, Dysphorie oder einer gescheiterten Studie.
Arzneiziele bedeutet Rezeptoren, die für Intervention in Betracht gezogen werden, nicht garantierte Erfolgsgeschichten. Eine selektive CB1-Ansprache könnte einige Off-Target-Effekte reduzieren, aber dennoch in zentrale Nebenwirkungen geraten. Eine selektive CB2-Ansprache kann einige intoxikationsbezogene Risiken vermeiden, doch Selektivität garantiert keine Wirksamkeit bei komplexen menschlichen Erkrankungen. Systembiologische Arbeiten machen das deutlich. Eine 2025/2026 auf PubMed indexierte integrative Netzwerkanalyse identifizierte CB1 und CB2 als besonders einflussreiche Knoten im Endocannabinoid-System und verknüpfte ihre Signalisierung mit breiteren metabolischen Wegen. Mit anderen Worten: Diese Rezeptoren liegen innerhalb größerer Netzwerke. Wenn man einen Knoten beeinflusst, bewegen sich andere Signalwege mit.
Das ist die Haltung dieses Artikels. CB1 und CB2 sind kontextabhängige Signal-Knoten. Keine statischen Schalter. Keine bloßen Etiketten für „Gehirn“ und „Immunsystem“. Wenn die cannabis-Wissenschaft erklären will, warum eine Verbindung in einem Kontext therapeutisch wirkt, in einem anderen intoxizierend ist und in einer Klinik enttäuscht, muss sie auf der Rezeptor-Ebene beginnen und dort lange genug bleiben, um der Biologie dorthin zu folgen, wo sie tatsächlich verläuft.
Eine kurze Geschichte der Entdeckung von Cannabinoid-Rezeptoren
Bevor Cannabinoid-Rezeptoren identifiziert wurden, war die cannabis Wissenschaft im Wesentlichen eine Geschichte der Chemie. Forschende konnten Pflanzenverbindungen isolieren, rohe Verhaltenswirkungen in Tieren vergleichen und über die Potenz diskutieren, aber sie konnten noch nicht erklären, wie ein Molekül wie Delta-9-Tetrahydrocannabinol oder THC seine Wirkungen mit auch nur annähernder Rezeptorpräzision hervorrief. Das änderte sich in den späten 1980er- und frühen 1990er-Jahren. Der Wandel war entscheidend: Die cannabis Forschung verlagerte sich vom Katalogisieren von Phytocannabinoiden hin zur Untersuchung von Ligand-Rezeptor-Interaktionen, intrazellulärer Signalübertragung, Gewebeverteilung und schließlich des endogenen Lipidsystems, das heute Endocannabinoid-System oder ECS genannt wird.
| Jahr | Meilenstein | Im Artikel genannte Personen |
|---|---|---|
| 1964 | Isolierung und Struktur von THC | Raphael Mechoulam; Yechiel Gaoni |
| 1988 | Spezifische hochaffine Cannabinoid-Bindestellen in Hirnmembranen der Ratte | Allyn Howlett; William Devane |
| 1990 | Klonierung von CB1 | Lisa Matsuda und Kolleginnen und Kollegen |
| 1992 | Identifizierung von Anandamid | William Devane; Lumír Hanuš; Raphael Mechoulam; Kolleginnen und Kollegen |
| 1993 | Identifizierung von CB2 | Sean Munro; Kerrie Thomas; M. Abu-Shaar |
| 1995 | Identifizierung von 2-AG durch getrennte Gruppen | Mechoulam-Team; Sugiura-Gruppe |
Von der THC-Pharmakologie zur Rezeptoridentifikation
Ein wichtiger früher Meilenstein kam 1964, als Raphael Mechoulam und Yechiel Gaoni die Isolierung und Struktur von THC berichteten. Diese Leistung war bedeutsam, weil sie Pharmakologinnen und Pharmakologen ein definiertes Molekül zum Testen gab statt eines variablen botanischen Extrakts. In den folgenden zwei Jahrzehnten erarbeitete das Fachgebiet eine Struktur-Wirkungs-Karte von THC und verwandten Cannabinoiden, doch über den Mechanismus wurde weiterhin gestritten. Einige Forschende bevorzugten unspezifische Membraneffekte, weil Cannabinoid lipophil sind. Diese Sichtweise war schwerer aufrechtzuerhalten, als Daten zu stereoselektiver und sättigbarer Bindung zunahmen.
Die Rezeptor-Ära begann eigentlich mit Bindungsstudien in den 1980er-Jahren. 1988 veröffentlichten Allyn Howlett und William Devane eine wegweisende Arbeit in Molecular Pharmacology, die spezifische Cannabinoid-Bindungsstellen mit hoher Affinität in Gehirnmembranen von Ratten unter Verwendung des synthetischen Agonisten CP55,940 zeigte. Das Ergebnis war kein vager Hinweis auf ein Ziel. Es zeigte Sättigbarkeit, regionale Variation und pharmakologische Spezifität, die mit einem echten Rezeptor vereinbar waren. Hirngewebe reagierte nicht auf Cannabinoid, als würden sie sich einfach in Lipid-Doppelschichten auflösen und alles zugleich stören. Es gab Selektivität.
Drei Jahre später, 1990, klonten Lisa Matsuda und Kolleginnen und Kollegen den ersten Cannabinoid-Rezeptor, heute CB1 genannt, und veröffentlichten ihn in Nature. CB1 wurde als G-Protein-gekoppelter Rezeptor oder GPCR identifiziert, was die Cannabinoid-Pharmakologie sofort in eine der wichtigsten Signaltransduktions-Superfamilien der Biologie einordnete. Das war bedeutsam, weil GPCRs nicht nur Schalter sind. Sie nehmen mehrere Konformationszustände an, koppeln an verschiedene intrazelluläre Partner, desensibilisieren, internalisieren und zeigen ligandenabhängige Signal-Bias. Diese Konzepte wurden erst viel später zentral, aber die Klonierung von CB1 machte sie überhaupt erst möglich.
CB2 folgte kurz darauf. 1993 identifizierten Sean Munro, Kerrie Thomas und M. Abu-Shaar einen zweiten Cannabinoid-Rezeptor, CB2, ebenfalls in Nature, zunächst charakterisiert aus immunbezogenen Geweben. Diese Entdeckung schuf eine dauerhafte Kurzform, die das Fachgebiet über Jahre prägte: CB1 als „Gehirn-Rezeptor“, verbunden mit Intoxikation, CB2 als „peripherer“ oder Immun-Rezeptor, verbunden mit Entzündung. Diese Kurzform war nützlich, aber selbst damals schon zu grob, und sie hat sich schlecht gehalten. Die Verteilung beider Rezeptoren hängt von Spezies, Zelltyp, Aktivierungszustand, Krankheitskontext und Testmethode ab.
Wie CB1 und CB2 das Endocannabinoid-Feld veränderten
Sobald CB1 und CB2 identifiziert waren, stellte sich die naheliegende nächste Frage: Warum besitzt der Körper überhaupt Rezeptoren für pflanzliche Cannabinoid? Die Antwort kam 1992, als William Devane, Lumír Hanuš, Raphael Mechoulam und Kolleginnen und Kollegen Anandamid, formal Arachidonoyl-Ethanolamid, als endogenen Liganden identifizierten. Die in Science veröffentlichte Arbeit markierte einen konzeptionellen Bruch. Die cannabis Pharmakologie handelte nicht mehr nur von exogenen Verbindungen aus Cannabis sativa. Sie handelte von einem körpereigenen Lipid-Signalsystem.
Ein zweiter wichtiger endogener Ligand, 2-Arachidonoylglycerol oder 2-AG, wurde 1995 von getrennten Gruppen identifiziert, darunter das Team von Mechoulam und die Gruppe von Sugiura. Mit Rezeptoren und endogenen Liganden an ihrem Platz expandierte das ECS rasch. Forschende identifizierten synthetisierende und abbauende Enzyme wie die Fettsäureamid-Hydrolase, FAAH, für Anandamid und die Monoacylglycerol-Lipase, MAGL, für 2-AG. Sie stießen auch auf eine weiterhin ungelöste Frage: wie diese stark lipophilen Moleküle durch Membranen und den extrazellulären Raum wandern. Das Fachgebiet spricht häufig von „Transport“, aber ein einzelner spezifischer Endocannabinoid-Transporter ist bislang schwer fassbar geblieben.
An diesem Punkt hörte die Cannabinoid-Wissenschaft auf, eine Zwei-Rezeptor-Tabelle zu sein, und wurde zu einem Signalnetzwerk. CB1 und CB2 wurden mit Gi/o-Proteinen, der Hemmung der Adenylylcyclase, der Modulation von Calcium- und Kaliumkanälen sowie der Hemmung der Transmitterfreisetzung verknüpft. Doch die Geschichte blieb nicht so einfach. Rezeptoren konnten Beta-Arrestine rekrutieren, Desensibilisierung und Internalisierung durchlaufen und unterschiedlich auf Phytocannabinoide, Endocannabinoid und synthetische Liganden reagieren, selbst wenn diese Liganden nominell denselben Rezeptor ansteuerten. Die heutige GPCR-Sprache des biased agonism passt besonders gut zu Cannabinoid. Ein Artikel aus dem Jahr 2025 im American Journal of Psychiatry argumentierte, dass CB1-biased signaling eine plausible therapeutische Strategie für Schizophrenie sei, eine Erkrankung, von der laut WHO weltweit etwa 24 Millionen Menschen betroffen sind. Das ist ein weiter Weg von dem alten Bild von CB1 als bloßem Rezeptor, der erklärt, warum THC intoxiziert.
Auch die Geschichte von CB2 hat sich erweitert. Frühe Arbeiten verorteten ihn vor allem in Immungeweben, und das war grundsätzlich richtig, aber spätere Studien fanden CB2-Expression in Mikroglia und unter bestimmten Bedingungen auch in anderen Zellpopulationen des zentralen Nervensystems. Eine 2026 in Frontiers in Behavioral Neuroscience veröffentlichte Übersicht beschrieb „an update over the last 3 years“ und verknüpfte CB2-Signaling mit neuroinflammatorischen und neurodegenerativen Mechanismen, was deutlich machte, dass CB2 nicht als für das Gehirn irrelevant abgetan werden kann. Die aktuelle Strukturarbeit ist noch weiter gegangen. Eine 2026 in Frontiers in Chemical Biology erschienene Übersicht zu „CB1 and CB2“ betonte, dass Subtyp-Selektivität von strukturellen Unterschieden auf Rezeptorebene abhängt, die Bindung, Wirksamkeit und Regulation verändern. Eine aktuelle PubMed-indexierte Studie zur Subtyp-Selektivität argumentiert ebenfalls, dass Endocannabinoid-Selektivität dynamisch ist und durch konformationelles Verhalten statt durch ein einfaches Schloss-und-Schlüssel-Modell geprägt wird.
Zentrale Forschende und warum die Geschichte weiterhin wichtig ist
Drei Namen gehören in den Mittelpunkt dieser Geschichte. Raphael Mechoulam half, die chemische und biologische Grundlage der Cannabinoid-Wissenschaft zu definieren, von der THC-Strukturarbeit bis zur Entdeckung von Endocannabinoid. Lumír Hanuš war eine zentrale Figur bei der Identifizierung von Anandamid und späterer Endocannabinoid-Forschung. Allyn Howletts Rezeptor-Pharmakologie war entscheidend, um zu beweisen, dass Cannabinoid über spezifische Bindungsstellen und Signalmechanismen im Gehirn wirken. Ohne diese Arbeiten gäbe es das moderne ECS-Fachgebiet nicht.
Die Geschichte bleibt wichtig, weil alte Vereinfachungen aktuelle Debatten weiterhin verzerren. Im Jahr 2019 verwendeten schätzungsweise 200 Millionen Menschen weltweit, etwa 4 % der 15- bis 64-Jährigen, cannabis laut WHO. Gleichzeitig gibt die FDA an, dass sie ein aus cannabis gewonnenes Arzneimittel und drei mit cannabis zusammenhängende Arzneimittel zugelassen hat. Die öffentliche Exposition ist enorm. Die klinische Translation ist selektiv und schwierig. Die Rezeptorgeschichte erklärt warum. Cannabinoid-Wirkungen hängen von Ligandenklasse, Rezeptorzustand, Gewebelokalisation, Zeitpunkt und Pathway-Bias ab. Sie hängen auch von einem breiteren Netzwerk ab. Eine integrative Netzwerkanalyse aus den Jahren 2025/2026 identifizierte CB1 und CB2 als hoch einflussreiche Knoten, die mit Stoffwechselwegen verbunden sind und nicht als isolierte Ziele fungieren.
Das ist das eigentliche Vermächtnis der Rezeptorentdeckung. Sie vereinfachte die Biologie von cannabis nicht. Sie zeigte, warum die Biologie komplizierter ist, als es die alte Trennung in Gehirn versus Körper je zuließ.
Wo CB1 vorkommt: Hirnschaltkreise, periphere Gewebe und funktionelle Gradienten
CB1 verdiente sich seinen Ruf als wichtiger psychoaktiver Cannabinoid-Rezeptor aus gutem Grund. Er ist im zentralen Nervensystem reichlich vorhanden, und Allyn Howletts Arbeit zur Rezeptor-Pharmakologie trug dazu bei, zu etablieren, dass THC über ein spezifisches, sättigbares Rezeptorsystem wirkt und nicht über unspezifische Membraneffekte. Aber die alte Kurzform — CB1 im Gehirn, CB2 in Immunzellen — stiftet inzwischen mehr Verwirrung als Klarheit. CB1 ist zwar stark in neuronalen Schaltkreisen angereichert. Er kommt aber auch im Darm, in der Leber, im Fettgewebe, in den Fortpflanzungsorganen, im kardiovaskulären Gewebe und in sensorischen Bahnen vor, wo er Nahrungsaufnahme, Stoffwechsel, Schmerzsignalgebung und autonome Funktion beeinflusst. Die Verteilung ist breit. Die Funktion ist kontextabhängig.
Das ist wichtig, weil die Exposition gegenüber Cannabinoid häufig ist. Die Weltgesundheitsorganisation schätzte, dass 2019 200 Millionen Menschen cannabis verwendeten, etwa 4 % der Weltbevölkerung im Alter von 15 bis 64 Jahren. Es ist auch wichtig, weil Rezeptor-Pharmakologie immer weiter in die Medizin hineinreicht: Die U.S. FDA gibt an, dass ein aus cannabis gewonnenes Arzneimittel und drei mit cannabis zusammenhängende Arzneimittel zugelassen sind. Ein Rezeptor, der in so vielen Organen vorkommt, lässt sich nicht auf ein einziges Verhaltenslabel reduzieren.
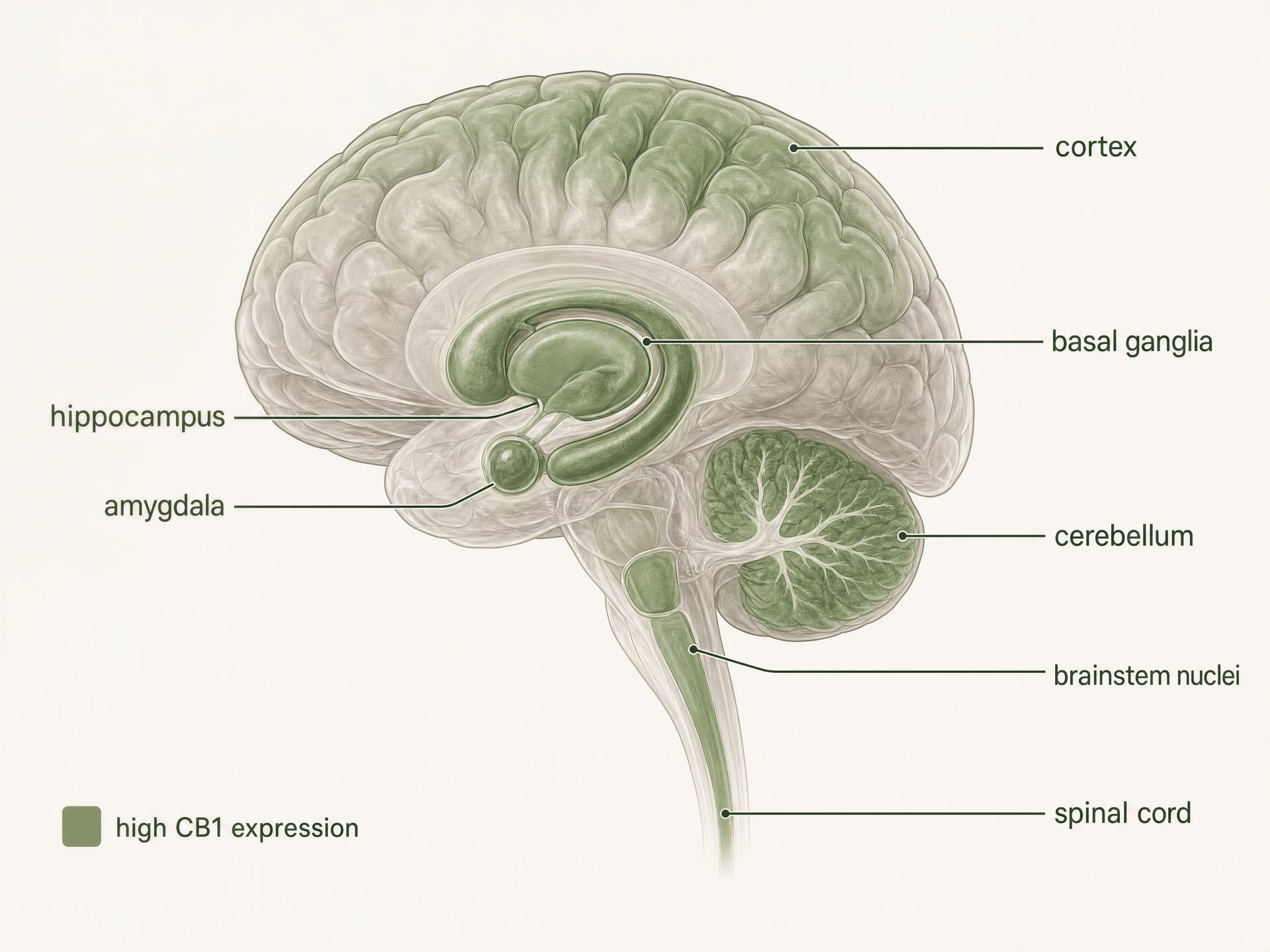
- Allgemeines Muster
- Einer der am stärksten vertretenen GPCRs im Säugetiergehirn
- Genannte Regionen mit hoher Dichte
- Kortex, Hippocampus, Amygdala, Basalganglien, Kleinhirn
- Genannte schmerzbezogene Orte
- Periaquäduktales Grau, rostrale ventromediale Medulla, Hinterhorn
- Genannte periphere Orte
- Darm, Leber, Fettgewebe, Fortpflanzung, kardiovaskuläre und sensorische Bahnen
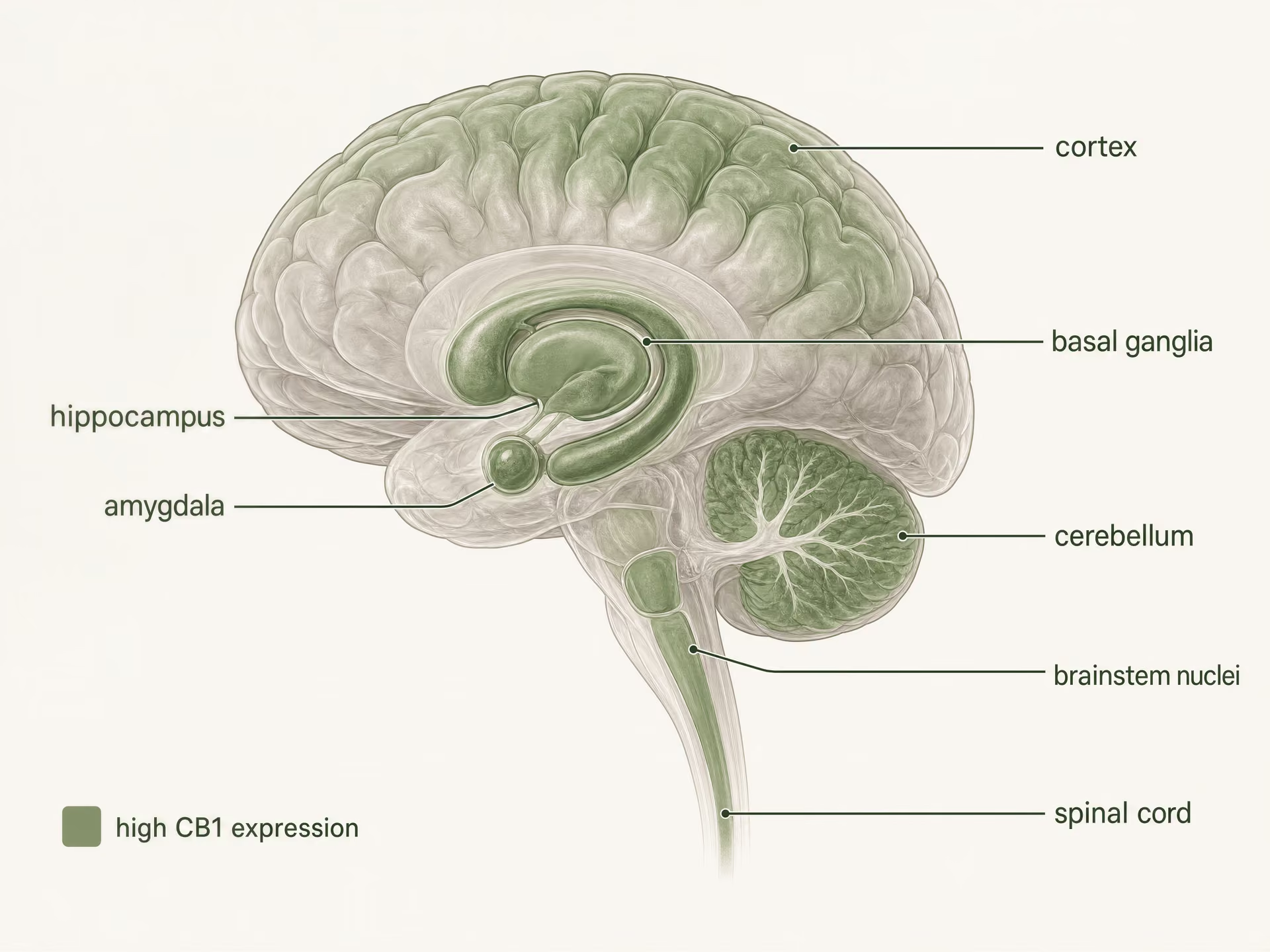
Hohe Expression im zentralen Nervensystem
{kind=link}
CB1 ist einer der am häufigsten vorkommenden G-Protein-gekoppelten Rezeptoren im Säugetiergehirn. Autoradiographie, In-situ-Hybridisierung und immunhistochemische Kartierungen zeichneten lange vor aktuellen Strukturstudien ein klares Bild: hohe Dichten finden sich in Kortex, Hippocampus, Amygdala, Basalganglien, Kleinhirn und mehreren schmerzverarbeitenden Regionen, mit zusätzlicher Expression in Hirnstammkernen und im gesamten Rückenmark. Dieses Muster passt überraschend gut, aber nicht perfekt, zu den klassischen Wirkungen von THC.
| CB1-Lokalisation | Unmittelbare synaptische Wirkung | Im Artikel genannter Beispiel-Effekt |
|---|---|---|
| GABAerge Interneuronen-Terminals | Unterdrückt GABA-Freisetzung | Enthemmung nachgeschalteter Neuronen |
| Glutamaterge Terminals | Unterdrückt Glutamatfreisetzung | Gedämpfte Erregung |
| Basalganglien- und Kleinhirnschaltkreise | Verändert die Transmitterfreisetzung in motorischen Bahnen | Verlangsamung der Motorik, veränderte Habitusschaltungen, beeinträchtigte Koordination |
| Schmerzbahnen | Moduliert nozizeptive Übertragung | Veränderungen der aufsteigenden, absteigenden, inflammatorischen und affektiven Schmerzverarbeitung |
Im Kortex und Hippocampus sitzt CB1 in Schaltkreisen, die Aufmerksamkeit, Arbeitsgedächtnis, Extinktionslernen und kurzfristige synaptische Plastizität regulieren. Gedächtniseffekte bedeuten nicht einfach nur „Hippocampus gleich Vergesslichkeit“. Sie hängen stark davon ab, welche Axonenden den Rezeptor exprimieren. CB1 ist oft präsynaptisch konzentriert, wo er nach Aktivierung durch Endocannabinoid wie Anandamid und 2-Arachidonoylglycerol die Neurotransmitterfreisetzung hemmt, jene Signallipide, deren Entdeckung durch Raphael Mechoulam, Lumír Hanuš und Kollegen das Feld grundlegend verändert hat. Wenn CB1 an Terminalen GABAerger Interneurone aktiviert wird, kann er nachgeschaltete Neurone enthemmen; wenn er an glutamatergen Terminalen aktiviert wird, kann er die Erregung dämpfen. Derselbe Rezeptor, gegenteiliges Netzwerkergebnis.
Die Basalganglien und das Kleinhirn erklären eine weitere vertraute Reihe von Effekten. Eine dichte CB1-Expression im Striatum, Globus pallidus, in der Substantia nigra pars reticulata und in den molekularen Schichten des Kleinhirns verknüpft die Rezeptoraktivierung mit motorischer Verlangsamung, veränderter Habit-Schaltkreisfunktion, beeinträchtigter Koordination und, bei einigen Dosen, katalepsieähnlichen Effekten in Tiermodellen. Doch die Tatsache, dass CB1 in den kardiorespiratorischen Zentren des Hirnstamms im Vergleich zu Opioidrezeptoren spärlich vertreten ist, hilft zu erklären, warum eine Überdosierung mit cannabinoid normalerweise nicht das gleiche Muster einer tödlichen Atemdepression hervorruft wie starke Opioidagonisten. Der Ort ist entscheidend. Ebenso das, was fehlt.[5]The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research. National Academies of Sciences, Engineering, and Medicine. National Academies Press, 2017. https://nap.nationalacademies.org/catalog/24625/the-health-effects-of-cannabis-and-cannabinoids-the-current-state
Die Schmerzverarbeitung zeigt dieselbe regionale Logik. CB1 findet sich in der periaquäduktalen Grauen Substanz, der rostralen ventromedialen Medulla, dem Hinterhorn des Rückenmarks und peripheren nozizeptiven Bahnen. Das verschafft dem Rezeptor mehrere Eintrittspforten in die Nozizeption: Er kann aufsteigende Schmerzsignale, die absteigende Schmerzkontrolle, inflammatorische Sensibilisierung und die emotionale Färbung von Schmerz beeinflussen. Das ist ein Grund dafür, warum cannabinoids in der Diskussion um chronische Schmerzen weiterhin eine Rolle spielen, zumal nach Angaben der National Academies fast 1 von 5 Erwachsenen in den Vereinigten Staaten mit chronischen Schmerzen lebt. Doch Analgesie ist nicht allein deshalb garantiert, weil CB1 vorhanden ist. Sedierung, kognitive Beeinträchtigung, Toleranz und dosislimitierende unerwünschte Wirkungen treten oft über benachbarte Schaltkreise oder über dieselben Schaltkreise bei unterschiedlichen Graden der Rezeptoraktivierung auf.
CB1-verzerrte Signalisierung könnte erwünschte therapeutische Wirkungen von unerwünschten psychoaktiven oder kognitiven Wirkungen trennen.Limited evidence
Gi/o-Proteine Eine Familie von G-Proteinen, die nach GPCR-Aktivierung typischerweise die Adenylylcyclase-Aktivität verringern und zur Steuerung von Ionenkanälen beitragen.
Die moderne Rezeptorbiologie fügt eine weitere Ebene hinzu. CB1 ist kein einfacher Ein-Aus-Schalter. Er koppelt vor allem an Gi/o-Proteine, reduziert die Adenylylcyclase-Aktivität und moduliert Ionenkanäle, kann aber auch Beta-Arrestine rekrutieren, Desensibilisierung und Internalisierung durchlaufen und ligandabhängige Signaling-Bias zeigen. Der 2025 im American Journal of Psychiatry erschienene Artikel, der argumentiert, dass sich ein biased signaling von CB1 therapeutisch bei Schizophrenie nutzen ließe, macht diesen Punkt direkt deutlich: Die reine Rezeptorbelegung ist ein schlechter Prädiktor für das Ergebnis. Da Schizophrenie weltweit etwa 24 Millionen Menschen betrifft, ist die Idee, erwünschte Signalwege von unerwünschten psychoaktiven oder kognitiven Effekten zu trennen, offensichtlich attraktiv. Ob diese Trennung praktisch erreichbar ist, bleibt eine offene Frage der Wirkstoffentwicklung, nicht eine feststehende Tatsache.
Peripheres CB1 in Darm, Leber, Fettgewebe und darüber hinaus
CB1 außerhalb des Gehirns ist keine Fußnote. Er ist zentral dafür, warum cannabinoids Appetit, Übelkeit, Glukoseverarbeitung, Lipidstoffwechsel und viszerale Wahrnehmung beeinflussen.
Im Darm wird CB1 im enterischen Nervensystem, in epithelialen Kompartimenten und in vagusbezogenen Bahnen exprimiert. Seine Aktivierung kann die Magenentleerung verlangsamen, die intestinale Motilität verändern, Emesis reduzieren und die Signalübertragung zwischen Darm und Gehirn verändern. Appetitwirkungen werden oft so beschrieben, als entstünden sie ausschließlich in hypothalamischen Belohnungs- und Fütterungszentren, doch peripheres CB1 trägt zur Gesamterscheinung bei, indem es sensorische und hormonelle Eingänge formt, bevor Signale diese Schaltkreise überhaupt erreichen. Eine Mahlzeit wirkt nicht auf eine leere Rezeptorlandschaft; sie verändert lokal den Endocannabinoid-Tonus.
In Leber und Fettgewebe ist CB1 an der metabolischen Regulation beteiligt, einschließlich Lipogenese, Insulinsensitivität und Energiespeicherung. Das war eine der wichtigsten Lehren der Rimonabant-Ära. Die Blockade von CB1 verbesserte Gewicht und metabolische Marker, was die Idee stützte, dass eine überaktive Endocannabinoid-Signalgebung zur Pathologie der Adipositas beiträgt. Rimonabant, ein zentral wirksamer CB1-Inversagonist, verursachte jedoch auch schwere psychiatrische unerwünschte Wirkungen, einschließlich Depression und Angst, und wurde vom Markt genommen. Diese Episode wird oft als Scheitern des „CB1-Targetings“ angeführt. Präziser war es das Scheitern einer bestimmten Art des CB1-Targetings: eine starke zentrale Antagonisierung oder Inversagonisierung in einem Rezeptorsystem, das in Stimmungs- und Stressschaltkreise eingebettet ist. Die Lehre lautet nicht, dass peripheres CB1 irrelevant ist; sie lautet, dass Expositionsmuster des Arzneimittels und Rezeptorzustand ebenso wichtig sind wie der Rezeptorname.
Adipozyten, Hepatozyten, pankreatisches Gewebe, Skelettmuskel, kardiovaskuläre Gewebe und Fortpflanzungsorgane erweitern die periphere Karte. Dasselbe gilt für sensorische Neurone. Die 2025/2026 in PubMed indexierte integrative Netzwerkanalyse, die CB1 und CB2 als hoch einflussreiche Knoten im Endocannabinoid-Signalnetzwerk identifizierte, ist hier nützlich, weil sie den Blick von der reinen Rezeptorlage auf die Beteiligung des Rezeptors an metabolischen und Signalnetzwerken verschiebt. Ein Rezeptor mit mäßiger Expression in einem Gewebe kann dennoch starke systemische Effekte haben, wenn er an einer Engstelle der lokalen Signalübertragung sitzt.
Auch strukturelle Arbeiten halten diese Diskussion nüchtern. Die 2026 in Frontiers in Chemical Biology erschienene Übersicht zu CB1 und CB2 betont, dass Ligandenselektivität und Wirksamkeit aus strukturellen Unterschieden auf Rezeptorebene entstehen, die Bindung, Signalübertragung und Rezeptorregulation verändern. Eine 2025/2026 in PubMed indexierte Studie zur Subtypselektivität argumentiert ebenfalls, dass Konformationsdynamik, nicht nur das Schloss-und-Schlüssel-Modell, bestimmt, wie Endocannabinoid-Rezeptorsubtypen unterscheiden. Das ist für die CB1-Verteilung wichtig, weil „CB1 in der Leber“ nicht bedeutet, dass THC, Anandamid, 2-AG und ein synthetischer Agonist dort alle dasselbe bewirken.
Warum Verteilung nicht mit einer einzigen einheitlichen Funktion gleichzusetzen ist
Der größte Fehler bei Rezeptorkarten besteht darin, Expression mit Schicksal gleichzusetzen. Das ist sie nicht. Hohe Expression sagt, wo man hinschauen muss, nicht, was passieren wird.
Erstens verändert der Zelltyp das Vorzeichen des Effekts. Ein CB1-Rezeptor an einem glutamatergen Endknöpfchen kann Erregung reduzieren. Derselbe Rezeptor an einem GABAergen Endknöpfchen kann Hemmung reduzieren. Das sind keine austauschbaren Ergebnisse. Zweitens ist die synaptische Lokalisation wichtig. CB1 sitzt meist präsynaptisch und wird oft durch Endocannabinoid aktiviert, die „on demand“ aus postsynaptischen Neuronen freigesetzt werden, wodurch eine retrograde Kontrolle der Neurotransmitterfreisetzung entsteht. Diese Anordnung begünstigt eine kurze, aktivitätsabhängige Modulation statt einer dauerhaften Rezeptoraktivierung.
Drittens ist die Ligandenidentität entscheidend. Endocannabinoid sind kurzlebige lokale Botenstoffe. Phytocannabinoid wie THC gelangen von außerhalb des Systems in den Organismus, oft in höheren und länger anhaltenden Expositionen als endogene Signale. Synthetische Liganden können noch stärker eingreifen, mit unterschiedlicher Wirksamkeit und unterschiedlichem Bias. Einige fördern Gi/o-Signalgebung stärker; andere begünstigen Beta-Arrestin-Rekrutierung, Desensibilisierung oder Rezeptorinternalisierung. Deshalb können zwei Verbindungen beide als CB1-Agonisten bezeichnet werden und sich dennoch deutlich in Appetitsteigerung, Gedächtnisstörung, motorischer Beeinträchtigung und Toleranzentwicklung unterscheiden.
Viertens verändert die lokale Ligandenverfügbarkeit alles. Anandamid und 2-AG werden vor Ort gebildet und abgebaut, sodass ihre Effekte von neuronaler Aktivität, metabolischem Zustand, Enzymexpression und inflammatorischem Kontext abhängen. Fünftens existiert auch die Rezeptordichte auf einem Gradienten. Hirnregion, Entwicklungsstadium, Krankheitszustand und wiederholte Arzneimittelexposition verschieben alle CB1-Niveaus und die Reaktionsfähigkeit.
Die aktuelle Literatur bewegt sich genau aus diesem Grund von binären Denkweisen weg. Die 2026 in Frontiers in Behavioral Neuroscience erschienene Übersicht erwähnt ein Update der letzten 3 Jahre darin, wie cannabinoid-Rezeptorsignalisierung bei Erkrankungen des ZNS verstanden wird, insbesondere sobald neuroinflammatorische und neurodegenerative Mechanismen einbezogen werden. CB1 sollte mit derselben Vorsicht gelesen werden. Er ist ein dominanter zentraler Rezeptor, aber nicht ausschließlich zentral; ein Fütterungsrezeptor, aber nicht nur das; ein Schmerzziel, aber kein sauberer Analgesieschalter. Jeder ernsthafte Bericht über die CB1-Verteilung muss in Gradienten, Schaltkreisen und Signalzuständen denken und nicht in einer Karikatur von Gehirn gegen Körper.
Wo CB2 gefunden wird: Wurzeln im Immunsystem und die wachsende ZNS-Karte
Die alte Kurzformel lautete klar: CB1 ist der Gehirnrezeptor, CB2 ist der Immunrezeptor. Diese Einordnung half in der frühen Lehre, führt heute jedoch eher in die Irre, als dass sie klärt. CB2 zeigt tatsächlich eine klassische Anreicherung außerhalb von Neuronen, insbesondere in immunologischen und hämatopoetischen Linien, und diese Tatsache bleibt pharmakologisch wichtig. Die neuere Literatur, insbesondere die 2026 in Frontiers in Behavioral Neuroscience erschienene Übersicht, vertritt jedoch eine stärkere These: CB2 wird nun im Zusammenhang mit Erkrankungen des zentralen Nervensystems diskutiert, weil seine Expression und Signalgebung in Mikroglia, inflammatorischen Schaltkreisen und verletzungsassoziierten Zuständen sichtbarer werden, ein „Update über die letzten 3 Jahre“, das die Kartierung und Interpretation des Rezeptors verändert hat. Das Ergebnis ist nicht, dass CB2 plötzlich ein hochabundanter, pan-neuronaler Gehirnrezeptor geworden wäre. Das ist er nicht. Das Ergebnis ist, dass die Verteilung des Rezeptors als bedingt, zelltypspezifisch und zustandsabhängig beschrieben werden muss.
Diese Unterscheidung ist klinisch wichtig. Die World Health Organization schätzte, dass 200 Millionen Menschen im Jahr 2019 cannabis konsumierten, also 4 % der Weltbevölkerung im Alter von 15–64 Jahren. Selbst bei nur einer kleinen Zahl zugelassener cannabinoid-bezogener Arzneimittel – die FDA zählte 2025 ein cannabis-abgeleitetes Produkt und drei cannabis-bezogene Produkte – bestimmt die Rezeptorlokalisation weiterhin, wo Wirkstoffentwickler nach antiinflammatorischen, analgetischen, neuroprotektiven und psychiatrischen Effekten suchen und wo sie unerwünschte Wirkungen erwarten.
- Klassische Anreicherung
- Immun- und hämatopoetische Zellen
- Genannte Zelltypen
- B-Zellen, T-Zellen, Makrophagen, Monozyten, natürliche Killerzellen, neutrophile Granulozyten, Mastzellen
- Genannte kanonische Gewebe
- Milz, Tonsille, Thymus, Knochenmark, zirkulierende Immunzellen
- Betonte ZNS-Relevanz
- Mikroglia und pathologiebezogene Zustände
Klassische Anreicherung in Immun- und Hämatopoetischen Zellen
CB2 wurde ursprünglich als der cannabinoid receptor-Subtyp mit der stärksten Expression in Zellen identifiziert, die eher mit Immunität als mit schneller synaptischer Übertragung verbunden sind. Das bleibt der richtige Ausgangspunkt. Im Vergleich zu CB1, der in vielen neuronalen Populationen stark vertreten ist, ist CB2 klassisch angereichert in B-Zellen, T-Zellen, Makrophagen, Monozyten, natural killer cells, Neutrophilen, Mastzellen und anderen hämatopoetischen Kompartimenten. Milz, Tonsillen, Thymus, Knochenmark und zirkulierende Immunzell-Populationen waren daher die kanonischen Gewebe für die CB2-Analyse.
Diese immunologische Gewichtung prägte die frühe Arzneimittelentwicklungs-Idee CB2-selektiver Agonisten als Möglichkeit, antiinflammatorische oder analgetische Vorteile zu nutzen und zugleich die mit einer starken CB1-Aktivierung im Gehirn verbundenen intoxicating effects zu vermeiden. Es war eine vernünftige Hypothese, aber nur halb vollständig. CB2 ist ein Gi/o-gekoppelter GPCR und schaltet wie CB1 nicht einfach nur „an“ oder „aus“. Abhängig von Ligand, Rezeptorkonformation und zellulärem Kontext kann CB2 die Adenylylcyclase-Aktivität senken, MAPK-Wege beeinflussen, die Ionenkanal-Kopplung indirekt verändern, beta-arrestins rekrutieren und Desensibilisierung oder Internalisierung durchlaufen. Selbst in peripheren Immungeweben lautet die eigentliche Frage also nicht nur, ob CB2 vorhanden ist, sondern welche Zellen es exprimieren, auf welchem Niveau, unter welchem Stimulus und mit welcher nachgeschalteten Bias.
Diese Komplexität ist einer der Gründe, warum ähnlich aussehende Liganden sich unterschiedlich verhalten können. Die 2026er Frontiers in Chemical Biology-Übersichtsarbeit zur Struktur des cannabinoid receptor argumentiert, dass die Selektivität an „CB1 und CB2“ durch strukturelle Unterschiede auf Rezeptorebene geprägt wird, die Ligandenbindung, Signalwirksamkeit und Rezeptorregulation verändern. Eine 2025/2026 in PubMed indexierte Studie zur Subtyp-Selektivität führte diesen Punkt weiter aus, indem sie zeigte, dass die endocannabinoid-Selektivität dynamisch ist und an Konformationszustände gebunden ist, nicht an ein starres Schlüssel-Schloss-Modell. Das ist für die Gewebekartierung wichtig, weil ein endogener Ligand wie 2-AG oder anandamide, ein phytocannabinoid wie THC und ein synthetischer CB2-bevorzugter Agonist zwar alle auf dieselbe Rezeptorpopulation treffen, aber unterschiedliche Signaloutputs stabilisieren können.
Die ältere, immunzentrierte Karte von CB2 war daher nicht falsch. Sie war unvollständig. CB2 lässt sich weiterhin am besten als ein Rezeptor mit starken Wurzeln im Immunsystem beschreiben. Aber Wurzeln sind nicht der ganze Organismus.
| Kontext | Wie CB2 beschrieben wird | Interpretationspunkt |
|---|---|---|
| Gesundes Hirnbasisniveau | In vielen Regionen oft niedrig oder nahe der Nachweisgrenze | Ein niedriges Basissignal bedeutet nicht Irrelevanz |
| Aktivierte Mikroglia | Nach Verletzung oder Entzündung besser nachweisbar | Stützt ZNS-Relevanz durch immunähnliche Funktionen |
| Astrozyten / Endothel / infiltrierende Zellen | In manchen Krankheitskontexten berichtet | Lokalisation hängt von Methode und Modell ab |
| Breite konstitutive neuronale Expression | Benötigt stärkere Evidenz | Der Artikel behandelt diese Behauptung mit Zurückhaltung |
CB2 in Mikroglia, Neuroinflammation und Verletzungszuständen
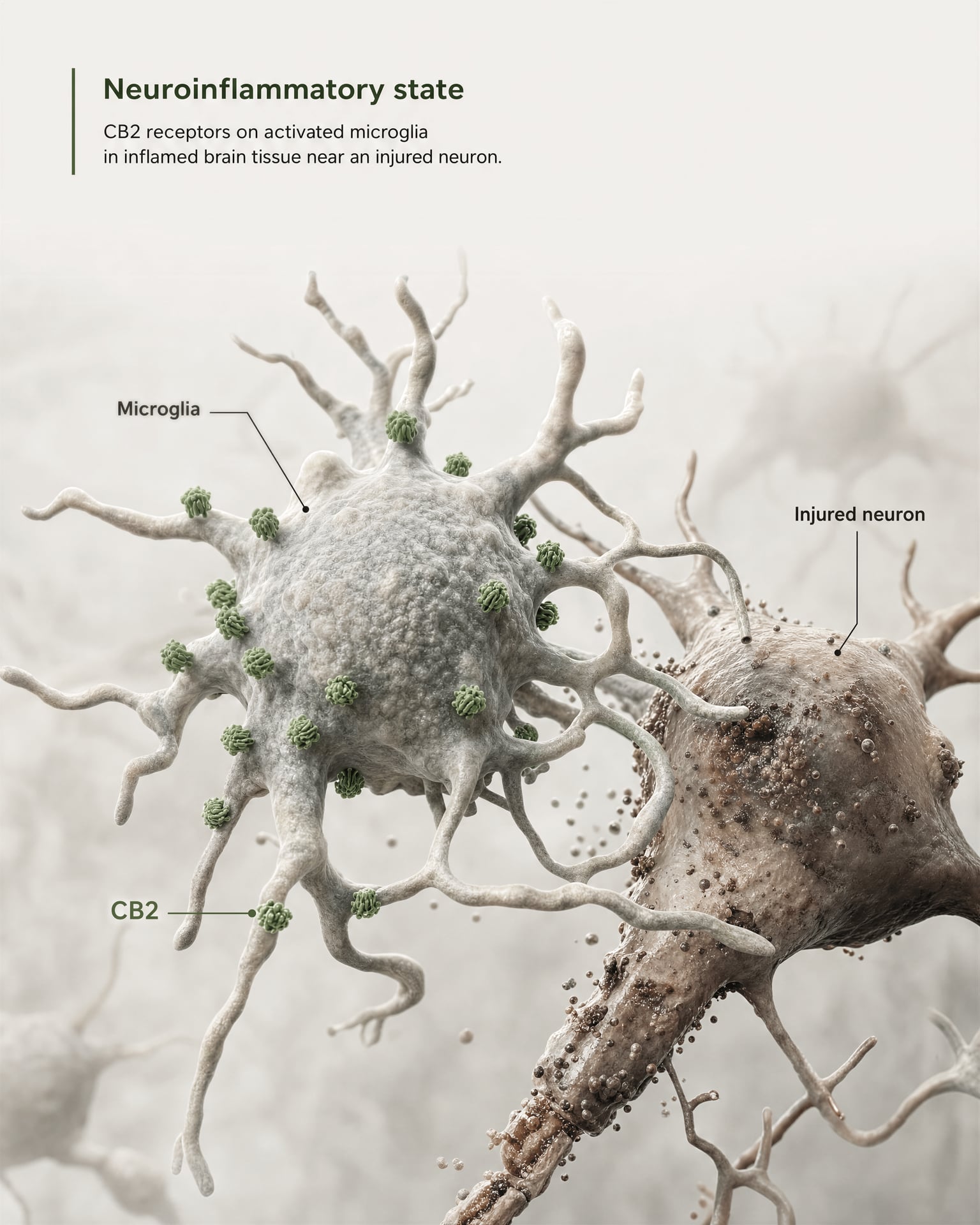
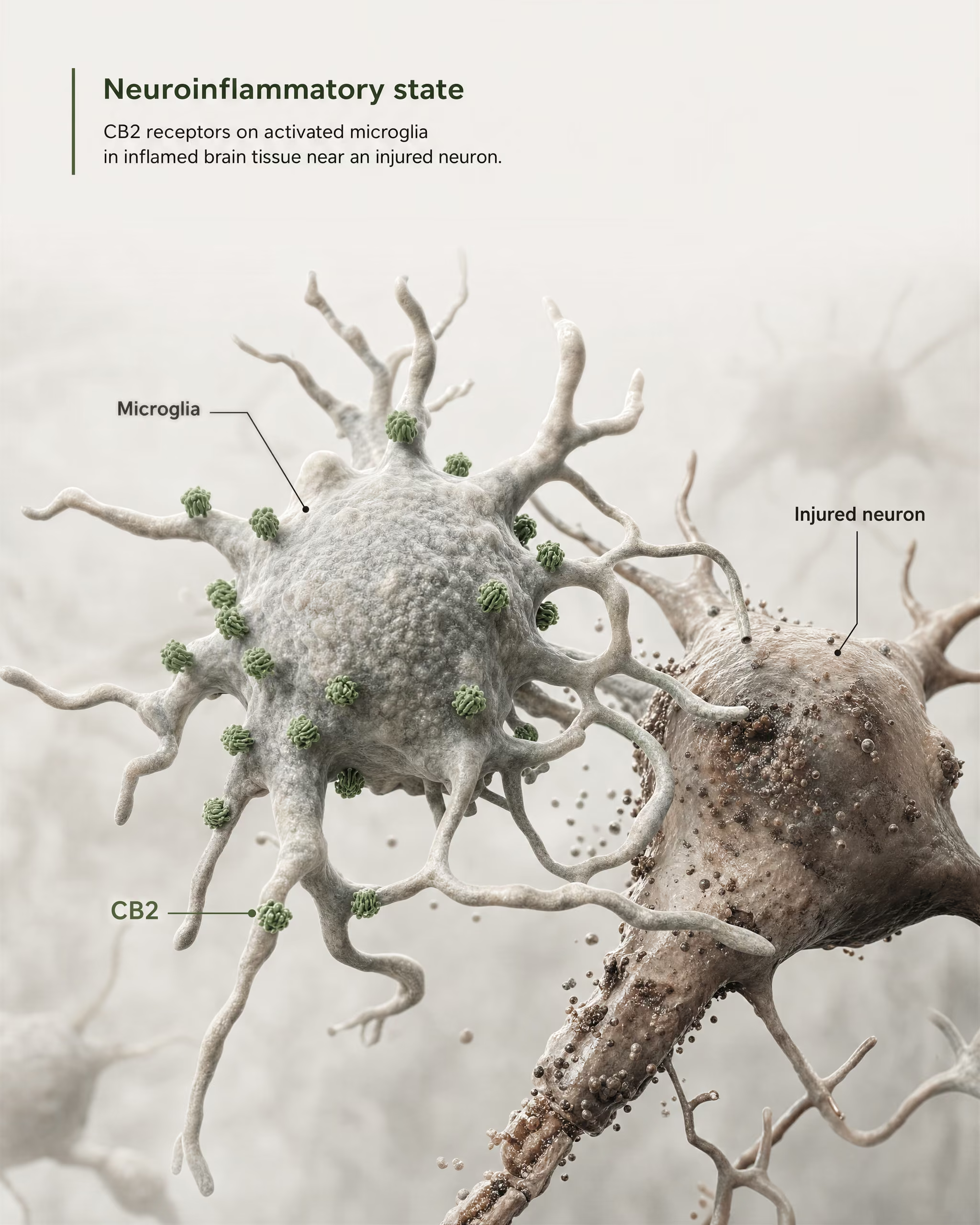
Das stärkste Argument für zentrale Relevanz ergibt sich nicht aus der Behauptung, CB2 sei auf gesunden Vorderhirn-Neuronen breit vorhanden. Es ergibt sich aus Mikroglia und aus der Krankheitsbiologie.
{kind=link}
Mikroglia sind die residenten Immunzellen des ZNS, und sie liegen genau an der Grenze, an der das alte Modell des „peripheren Immunrezeptors“ zu versagen beginnt. Wenn ein Rezeptor im eigenen immunologischen Überwachungssystem und in der inflammatorischen Antwort des Gehirns exprimiert wird, ist die Bezeichnung lediglich peripher ungenau. Die 2026er Frontiers in Behavioral Neuroscience-Übersichtsarbeit macht diesen Punkt direkt: CB2-Signaling hat Aufmerksamkeit in ZNS-Erkrankungen gewonnen, weil es mit neuroinflammatorischen und neurodegenerativen Mechanismen verknüpft ist. Deshalb erscheint CB2 heute in Diskussionen über Alzheimer-Krankheit, Parkinson-Krankheit, Multiple Sklerose, traumatische Hirnverletzung, Schlaganfall, neuropathischen Schmerz und einige psychiatrische Erkrankungen, bei denen inflammatorische Signale Teil der Pathologie sind.
Der Schlüsselbegriff lautet induzierte oder erhöhte Expression. In vielen gesunden Hirnregionen ist die basale CB2-Expression niedrig, manchmal nahe an den Nachweisgrenzen älterer Methoden. Nach Verletzung, Infektion, chronischer Inflammation oder Neurodegeneration wird das CB2-Signal jedoch oft besser nachweisbar, insbesondere in aktivierten Mikroglia und, in einigen Studien, auch in Astrozyten, infiltrierenden Immunzellen, endothelialen Kompartimenten oder begrenzten neuronalen Subsets. Das ist eine völlig andere Verteilungsregel als diejenige, die üblicherweise für CB1 gilt. CB1 ist häufig konstitutiv in definierten neuronalen Schaltkreisen reichlich vorhanden. CB2 wird eher als ein Rezeptor interpretiert, dessen ZNS-Relevanz unter Stress, Pathologie oder inflammatorischer Aktivierung sichtbar wird.
Diese Unterscheidung hat praktische Konsequenzen. Ein auf CB2 zielendes Medikament kann in gesundem Gewebe mit niedriger Rezeptordichte wenig Wirkung zeigen, in krankem Gewebe mit erhöhter Expression und veränderten Signalnetzwerken jedoch messbare Aktivität entfalten. Diese Induzierbarkeit ist einer der Gründe, warum präklinische Befunde sowohl spannend als auch schwer zu übertragen waren. Der Zeitpunkt zählt. Das Krankheitsstadium zählt. Die Zellzusammensetzung zählt. Ein mikrogliales Milieu nach einer Verletzung ist pharmakologisch nicht mit einem nicht stimulierten Hirnschnitt gleichzusetzen.
Die interpretativen Probleme sind nicht trivial. CB2 hat eine lange Geschichte von Problemen mit Antikörperspezifität, der Detektion niedrig abundanter Transkripte, Speziesunterschieden und inkonsistenten Lokalisationsansprüchen über verschiedene Methoden hinweg. Einige frühe Berichte haben neuronales CB2 vermutlich überschätzt, weil die verfügbaren Werkzeuge schwach waren. Deshalb stützen sich sorgfältige Studien heute auf konvergierende Evidenz—Single-Cell-Transkriptomik, in situ hybridization, validierte genetische Reporter, Knockout-Kontrollen, proteomische Daten, soweit möglich, und zustandsabhängige Vergleiche—statt auf ein einzelnes Färbeergebnis. Wenn eine Studie CB2 in Neuronen im Basalzustand berichtet und eine andere es nicht nachweisen kann, kann die Abweichung echte regionale Unterschiede, den Krankheitsstatus, die Spezies, das Alter oder schlicht Assay-Limitierungen widerspiegeln.
CB2 hat eine relevante ZNS-Bedeutung, insbesondere in glialen und verletzungsbezogenen inflammatorischen Kontexten.Limited evidence
Die beste aktuelle Position ist daher zurückhaltend, aber klar: CB2 hat eine reale ZNS-Relevanz, vor allem über gliale und immunähnliche Funktionen, und diese Relevanz nimmt bei Neuroinflammation und Verletzung zu. Behauptungen über eine breite konstitutive neuronale CB2-Expression im normalen Gehirn benötigen stärkere Evidenz als Behauptungen über mikrogliales und pathologieassoziiertes CB2.
Wie die letzten 3 Jahre die CB2-Debatte verändert haben
Die 2026er Frontiers in Behavioral Neuroscience-Übersichtsarbeit rahmt die neuere Literatur ausdrücklich als ein „update over the last 3 years“, und diese Formulierung fängt einen echten Wandel ein. Die Debatte verschob sich weg von der Frage, ob CB2 „überhaupt im Gehirn“ sei, hin zur Frage, wo, wann und in welchen Krankheitszuständen sein Signaling angreifbar wird.
Drei Entwicklungen trieben diesen Wandel voran. Erstens verbesserten sich Zellauflösungs-Methoden. Single-Cell- und Single-Nucleus-RNA-Datensätze, bessere räumliche Kartierung und strengere Validierungsstandards verringerten die Wahrscheinlichkeit, dass niedriggradige oder induzierbare Expression einfach verworfen wird, nur weil ältere Assays unempfindlich waren. Zweitens wurde Neuroinflammation für viele Modelle von Hirnerkrankungen zentral. Sobald Erkrankungen durch immunologische und gliale Mechanismen statt durch reine Neuron-Frameworks analysiert wurden, wurde CB2 deutlich schwerer zu ignorieren. Drittens reifte die Rezeptorpharmakologie selbst. Das Feld denkt heute stärker in Kategorien von Wirksamkeit, signal bias, Rezeptor-Trafficking und kontextabhängigen Antworten als in bloßer Belegung.
Diese breitere GPCR-Perspektive ist sogar außerhalb der CB2-Literatur sichtbar. Der 2025er American Journal of Psychiatry-Artikel über CB1 biased signaling und Schizophrenie argumentiert, dass cannabinoid pharmacology durch biased signaling und nicht durch grobe Rezeptoraktivierung verstanden werden sollte. Schizophrenie betrifft laut WHO weltweit etwa 24 Millionen Menschen, also ist dies keine akademische Randfrage. Dieselbe Logik gilt für CB2. Ein Ligand, der auf dem Papier „CB2-selektiv“ ist, kann dennoch unterschiedliche Ergebnisse erzeugen, je nachdem, ob er bevorzugt G-Protein-Signaling, beta-arrestin-Rekrutierung, Rezeptorinternalisierung oder antiinflammatorische transkriptionelle Programme in aktivierter Mikroglia auslöst.
Die neuere Systemperspektive stärkt dies zusätzlich. Eine 2025/2026 in PubMed indexierte Netzwerk-Analyse-Studie identifizierte CB1 und CB2 als besonders einflussreiche Knoten im endocannabinoid system und verknüpfte Rezeptor-Signaling mit metabolischen Wegen, statt Rezeptoren von der restlichen Zellbiologie zu isolieren. Das passt zu dem, was die ZNS-Daten zu CB2 zeigen: Verteilung ist kein fester Atlas-Eintrag. Sie ist Teil eines adaptiven Signalnetzwerks.
Die Schlussfolgerung ist einfach. CB2 sollte weiterhin als immunangereicherter cannabinoid receptor eingeführt werden. Dort zu stoppen vermittelt jedoch ein falsches Bild. Im Gehirn ist CB2 am besten als ein niedrig basal exprimierter, induzierbarer, krankheitsassoziierter Rezeptor zu verstehen, dessen Bedeutung in Mikroglia und neuroinflammatorischen Zuständen am klarsten wird—und dessen Nachweis weiterhin stark von Methode, Modell und Timing abhängt.
Wie cannabinoid receptors signalisieren: Gi/o-Kopplung, Second Messenger und synaptische Effekte
Die Pharmakologie der cannabinoid receptors beginnt mit einer einfachen Aussage, die sich rasch verkompliziert: CB1 und CB2 sind Class-A-G-Protein-gekoppelte Rezeptoren, und beide signalisieren am häufigsten über Gi/o-Proteine. Diese grundlegende Tatsache, die durch grundlegende Rezeptorarbeit von Allyn Howlett und anderen etabliert wurde, gilt weiterhin. Geändert hat sich das Verständnis dessen, was Gi/o-Kopplung in Zellen tatsächlich bedeutet. Sie bedeutet nicht einen einzigen nachgeschalteten Effekt. Sie bedeutet ein Spektrum möglicher Effekte, deren Zusammensetzung von Ligand, Rezeptordichte, Phosphorylierungszustand, Membranumgebung, Zelltyp und Zeitverlauf abhängt.
Diese Unterscheidung ist wichtig, weil laut der Weltgesundheitsorganisation im Jahr 2019 rund 200 Millionen Menschen cannabis konsumierten, also 4 % der Weltbevölkerung im Alter von 15 bis 64 Jahren, während die FDA angibt, dass mit Stand 2025 ein cannabis-abgeleitetes Arzneimittel und drei mit cannabis in Zusammenhang stehende Arzneimittel zugelassen sind. Rezeptorsignalisierung ist keine Nebensache. Sie ist der Mechanismus, der ein nützliches Antiseizure drug von Sedierung, ein gescheitertes Appetitanreger-Medikament von psychiatrischen Nebenwirkungen und einen im Labor selektiven Liganden von einem klinisch enttäuschenden unterscheidet.
Kanonische GPCR-Signalisierung bei CB1 und CB2
Kanonische CB1/CB2-Signalkaskade
- Ligandenbindung Ein Agonist stabilisiert eine aktive Rezeptorkonformation.
- G-Protein-Aktivierung Der Rezeptor fördert den GDP-GTP-Austausch an Gi/o.
- Untereinheiten-Trennung Galpha und Gbeta-gamma regulieren nachgeschaltete Effektoren.
- Veränderung des Second Messengers Die Adenylylcyclase-Aktivität sinkt und cAMP nimmt ab.
- Zelluläre Wirkung Ionenkanäle, Transmitterfreisetzung, Kinasen und Genregulation verschieben sich.
Im kanonischen Modell stabilisiert die Bindung eines Agonisten eine aktive Rezeptorkonformation, der Rezeptor wirkt als Guanin-Nukleotid-Austauschfaktor für Gi/o, Gαi/o tauscht GDP gegen GTP aus, und die Gα- und Gβγ-Komponenten regulieren anschließend nachgeschaltete Effektoren. Für CB1 und CB2 ist der klassische Readout die Hemmung der Adenylylcyclase und ein Abfall des intrazellulären zyklischen AMP. Dieser Befund wurde zu einer der frühesten biochemischen Signaturen, mit denen sich cannabinoid receptor activity definieren ließ.
„Kanonisch“ sollte jedoch nicht als „einheitlich“ gelesen werden. CB1 zeigt in mehreren Expressionssystemen eine hohe konstitutive Aktivität, was bedeutet, dass der Rezeptor auch ohne vorhandenen Agonisten messbar signalisieren kann. Diese Eigenschaft hilft zu erklären, warum Inverse Agonisten wie Rimonabant nicht nur den endogenen cannabinoid-Tonus blockierten, sondern die Signalisierung unter das Baseline-Niveau drückten und ausgeprägte zentrale Nebenwirkungen verursachten. Auch CB2 koppelt an Gi/o, doch die Art und Weise, wie Liganden aktive Zustände stabilisieren, unterscheidet sich von CB1. Strukturarbeiten, die 2026 in Frontiers in Chemical Biology besprochen wurden, betonten, dass Subtypselektivität zwischen „CB1 und CB2“ durch Unterschiede auf Rezeptorebene getrieben wird, die nicht nur die Bindung, sondern auch Wirksamkeit und Regulation verändern. Eine 2025/2026 in PubMed indexierte Studie zur Subtypselektivität ging noch weiter und argumentierte, dass endocannabinoid-Selektivität dynamisch ist und eher durch Konformationsverhalten als durch ein starres Schloss-Schlüssel-Modell geprägt wird.
Das ist einer der Gründe, warum phytocannabinoids, endocannabinoids und synthetische Liganden niemals als austauschbar behandelt werden sollten. Anandamid, identifiziert von Raphael Mechoulam und Lumír Hanuš, sowie 2-Arachidonylglycerol sind endogene Liganden, die bei Bedarf gebildet und rasch inaktiviert werden. Δ9-THC ist ein pflanzlicher Partialagonist mit Kinetik und Wirksamkeit, die sich von denen dieser endocannabinoids unterscheiden. Synthetische Agonisten wie CP55,940, WIN55,212-2 oder HU-210 treiben häufig eine stärkere Rezeptoraktivierung an und können Signalwege in unterschiedlichem Ausmaß rekrutieren. Manche Liganden begünstigen G-Protein-Signalisierung gegenüber β-Arrestin-Rekrutierung; andere nicht. Der Artikel im American Journal of Psychiatry aus dem Jahr 2025 machte diesen Punkt für CB1 direkt und argumentierte, dass biased signaling eine plausible therapeutische Strategie bei Schizophrenie ist, einer Erkrankung, von der weltweit etwa 24 Millionen Menschen betroffen sind.
CB2 ergänzt eine weitere Korrektur älterer Vereinfachungen. Es ist weiterhin in vielen Immunzellpopulationen angereichert, doch die 2026 in Frontiers in Behavioral Neuroscience erschienene Übersicht beschrieb „an update over the last 3 years“, in dem CB2-Signalisierung bei zentralnervösen Erkrankungen im Zusammenhang mit Neuroinflammation und Neurodegeneration mehr Aufmerksamkeit erhielt. Noch bevor also biased agonism ins Spiel kommt, scheitert die alte Zweiteilung in einen „Hirnrezeptor“ und einen „Immunrezeptor“ bereits auf der Ebene des Signalkontexts.
Effekte auf cAMP, Ionenkanäle und Neurotransmitterfreisetzung
Der bekannteste Second-Messenger-Effekt an beiden Rezeptoren ist die Unterdrückung der cAMP-Bildung durch Hemmung der Adenylylcyclase. Niedrigeres cAMP bedeutet oft niedrigere Aktivität der Proteinkinase A, veränderte Phosphorylierung nachgeschalteter Zielstrukturen und langsamere Änderungen der Genexpression über Signalwege wie CREB. In Neuronen sind jedoch häufig die schnellen Effekte wichtiger als die langsamen.
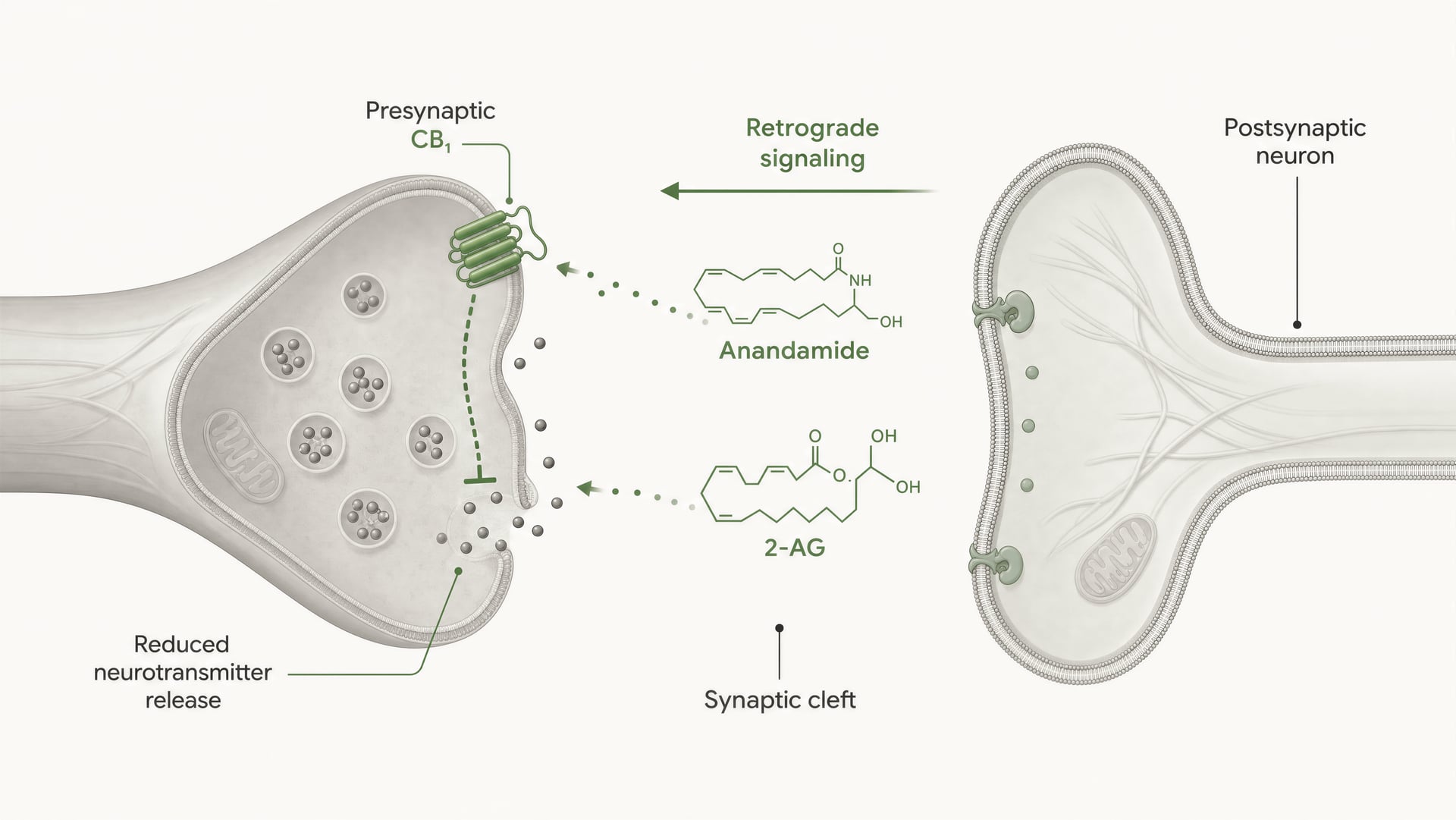
CB1 ist stark für die präsynaptische Kontrolle positioniert. In vielen Hirnschaltkreisen sitzt er auf Axonendigungen, wo die Rezeptoraktivierung die Wahrscheinlichkeit der Neurotransmitterfreisetzung reduziert. Dies geschieht durch eine Kombination aus Gβγ-vermittelter Hemmung spannungsabhängiger Calciumkanäle und Aktivierung von inwardly rectifying potassium conductances oder anderen Kaliumströmen, die die Erregbarkeit der Endigung dämpfen. Weniger Calciumeinstrom bedeutet, dass weniger Vesikel fusionieren. Das Ergebnis ist eine geringere Transmitterausschüttung in den synaptischen Spalt.
Das ist der Kernmechanismus der kurzreichweitigen retrograden endocannabinoid-Signalisierung. Ein postsynaptisches Neuron wird aktiv, synthetisiert endocannabinoids bei Bedarf aus membranständigen Lipidvorstufen und sendet sie rückwärts über die Synapse, um präsynaptische CB1-Rezeptoren zu aktivieren. Die präsynaptische Endigung setzt dann weniger Transmitter frei. Das ist eine Feedback-Bremse. An exzitatorischen Synapsen kann dies die Glutamatfreisetzung unterdrücken; an inhibitorischen Synapsen kann es die GABA-Freisetzung unterdrücken. Die Richtung des Netzwerkausgangs hängt davon ab, welche Endigung CB1 exprimiert. Derselbe Rezeptor, entgegengesetzte Netzwerkfolge.
| Begriff | Was unterdrückt wird | Beschriebener Mechanismus |
|---|---|---|
| DSI | Hemmung | Postsynaptische Aktivität setzt Endocannabinoide frei, die präsynaptisches CB1 aktivieren und die GABA-Freisetzung senken |
| DSE | Erregung | Postsynaptische Aktivität setzt Endocannabinoide frei, die präsynaptisches CB1 aktivieren und die Glutamatfreisetzung senken |
DSI und DSE Kurzfristige Formen endocannabinoidvermittelter synaptischer Plastizität, bei denen postsynaptische Depolarisation die inhibitorische Übertragung (DSI) oder die exzitatorische Übertragung (DSE) über präsynaptische CB1-Aktivierung unterdrückt.
Klassische physiologische Begriffe fassen dies zusammen: depolarization-induced suppression of inhibition, DSI, und depolarization-induced suppression of excitation, DSE. Beide sind kurzzeitige Formen synaptischer Plastizität, die durch endocannabinoid-Freisetzung und präsynaptische CB1-Aktivierung angetrieben werden. Auch länger anhaltende Effekte treten auf, einschließlich endocannabinoid-vermittelter Langzeitdepression an einigen Synapsen. Diese Phänomene sind wichtig, weil sie Rezeptorbiochemie mit Verhalten verbinden: Schmerzverarbeitung, Furchtextinktion, Gewohnheitslernen, Appetit, motorische Kontrolle und Anfallschwelle hängen alle von dieser Abstimmung der Freisetzungswahrscheinlichkeit ab.
Die Details sind nicht trivial. Ein Partialagonist wie Δ9-THC bildet möglicherweise nicht das vollständige Muster ab, das durch einen kurzen endogenen 2-AG-Impuls erzeugt wird. Ein synthetischer Vollagonist wird die physiologische Zeitgebung ebenfalls nicht notwendigerweise erhalten. Die Dosis ist wichtig. Ebenso die Rezeptorreserve. In einer Synapse mit dichter CB1-Expression kann selbst ein Partialagonist einen großen Effekt auf die Transmitterfreisetzung ausüben. In einem anderen Gewebe kann derselbe Ligand schwach erscheinen.
CB2 besitzt eine weniger gut etablierte direkte synaptische Physiologie als CB1, reduziert aber ebenfalls cAMP und kann Calcium-Signaling, Kinasewege und die Freisetzung inflammatorischer Mediatoren in Immun- und Gliazellen regulieren. Das macht CB2 relevant für die Neuron-Glia-Kommunikation, insbesondere in Krankheitszuständen, in denen sich die Rezeptorexpression verändert. Der in PubMed 2025/2026 indexierte Netzwerk-Analyse-Artikel behandelte CB1 und CB2 als einflussreiche Knoten im breiteren endocannabinoid- und metabolischen Signaling, was eine bessere Perspektive ist, als sie als isolierte Schalter zu betrachten.
Desensibilisierung, Internalisierung und Rezeptorregulation
Wie Rezeptoren auf wiederholte Agonistenexposition reagieren
- Phosphorylierung Intrazelluläre Rezeptorbereiche werden durch GPCR-Kinasen und andere Kinasen modifiziert.
- Beta-Arrestin-Rekrutierung Arrestine entkoppeln Rezeptoren von G-Proteinen und können zusätzliche Signale auslösen.
- Desensibilisierung Der Rezeptor reagiert weniger stark.
- Internalisierung Rezeptoren werden in endozytotische Wege aufgenommen.
- Schicksal nach der Aufnahme Rezeptoren können zur Oberfläche zurückkehren oder abgebaut werden.
Kein Rezeptor kann ohne Konsequenzen kontinuierlich aktiviert werden. Bei CB1 und CB2 führt eine prolongierte oder wiederholte Agonistenexposition häufig zu einer Phosphorylierung der intrazellulären Regionen des Rezeptors durch GPCR-Kinasen und andere Kinasen, zur Rekrutierung von β-Arrestinen, zur Entkopplung von G-Proteinen und anschließend zur Internalisierung über endozytotische Wege. Zunächst kommt die Desensibilisierung. Danach folgt oft die Endozytose. Recycling oder Abbau schließen sich an.
Für CB1 ist dieser Regulationszyklus ein wesentlicher Grund dafür, dass sich akute und chronische Effekte unterscheiden. Starke Agonisten können in Zellsystemen eine rasche Desensibilisierung und in vivo messbare Toleranz auslösen. Die regionsspezifische Regulation ist hier wichtig. CB1-Rezeptoren desensibilisieren nicht in allen neuronalen Populationen gleich, was hilft zu erklären, warum sich Toleranz über cannabinoid-Effekte hinweg ungleich entwickelt. Analgetische Reaktionen, Hypothermie, Gedächtnisstörungen und motorische Effekte können sich in unterschiedlichem Tempo verschieben, weil der Rezeptor in verschiedenen Schaltkreisen unterschiedlich reguliert wird.
β-Arrestine sind nicht nur Abschalter. Sie können eigene Signalkaskaden scaffolden, einschließlich MAP-Kinase-Signalwege, weshalb die Arrestin-Rekrutierung für biased agonism zentral geworden ist. Ein Ligand, der cAMP stark hemmt, aber β-Arrestin nur schwach rekrutiert, kann sich anders verhalten als einer, der beides effizient tut. Das ist längst kein theoretisches Detail mehr; es ist eine Strategie des Wirkstoffdesigns. Die 2025er Diskussion im American Journal of Psychiatry über CB1-Bias bei Schizophrenie spiegelt eine breitere GPCR-Lehre wider: Das Vermeiden bestimmter Signalarme kann einige Risiken reduzieren, doch Selektivität für einen Signalweg garantiert keinen klinischen Erfolg.
Auch die Internalisierung selbst ist ligandabhängig. Manche Agonisten treiben eine ausgedehnte Rezeptorendocytose an; andere bewirken trotz G-Protein-Aktivierung nur eine geringe Internalisierung. Allosterische Modulatoren verkomplizieren das Bild zusätzlich, indem sie verändern, wie orthosteric Ligands Rezeptorzustände stabilisieren. Hier trifft Strukturfarmakologie auf Therapeutik. Die 2026er Strukturübersicht machte deutlich, dass die Rezeptorkonformation Signalwirksamkeit und Rezeptorregulation gemeinsam steuert, nicht als getrennte Themen.
Das ist die zentrale Signallehre, die man mitnehmen sollte. CB1 und CB2 sind keine einfachen Ein-Aus-Detektoren für cannabinoids. Sie sind regulierte Knotenpunkte, deren Output sich über Millisekunden bis Tage verändert. Jeder ernsthafte Versuch, sie therapeutisch zu adressieren, sei es bei Epilepsie, Schmerzen, Psychose oder entzündlichen Erkrankungen, muss Gi/o-Kopplung, Second Messenger, Ionenkanalsteuerung, synaptische Lokalisation und die Tatsache berücksichtigen, dass der Rezeptor sich an anhaltenden Stimulus anpasst.
Biasedes Signaling: Warum Ein Rezeptor Unterschiedliche Biologische Ergebnisse Hervorrufen Kann
Das alte Bild der cannabinoid-Pharmakologie behandelte einen Rezeptor wie einen Lichtschalter: Agonisten schalten ihn ein, Antagonisten schalten ihn aus, und alles Weitere ergibt sich daraus, wo dieser Rezeptor zufällig exprimiert wird. Dieses Bild ist für CB1 oder CB2 nicht ausreichend. Es erklärt nicht, warum zwei Liganden, die am selben Rezeptor wirken, deutlich unterschiedliche Verhaltens-, kognitive, entzündliche oder therapeutische Effekte hervorrufen können. Es erklärt auch nicht, warum die Wirkstoffentwicklung rund um cannabinoid-Rezeptoren wiederholt Verbindungen hervorgebracht hat, die in vitro vielversprechend erschienen, sich jedoch als enttäuschend, schlecht verträglich oder klinisch uneindeutig erwiesen.
Das ist weit über die akademische Rezeptortheorie hinaus relevant. Die World Health Organization schätzte, dass im Jahr 2019 200 Millionen Menschen cannabis konsumierten, also etwa 4 % der Weltbevölkerung im Alter von 15 bis 64 Jahren. Schizophrenie betrifft weltweit etwa 24 Millionen Menschen. Vor diesem Hintergrund ist die Pharmakologie von CB1 keine Randfrage. Sie liegt an der Schnittstelle von öffentlicher Gesundheit, Psychiatrie und Wirkstoffdesign. Die U.S. FDA hatte bis 2025 ein cannabis-abgeleitetes Arzneimittel und drei cannabis-bezogene Arzneimittel zugelassen, eine kleine Zahl im Vergleich zum Ausmaß des klinischen Interesses. Ein Grund dafür, dass der Fortschritt langsamer verläuft als die öffentliche Diskussion vermuten lässt, ist, dass cannabinoid-Rezeptor-Signaling nicht einfach nur Rezeptorbelegung ist. Es ist Pfadauswahl.
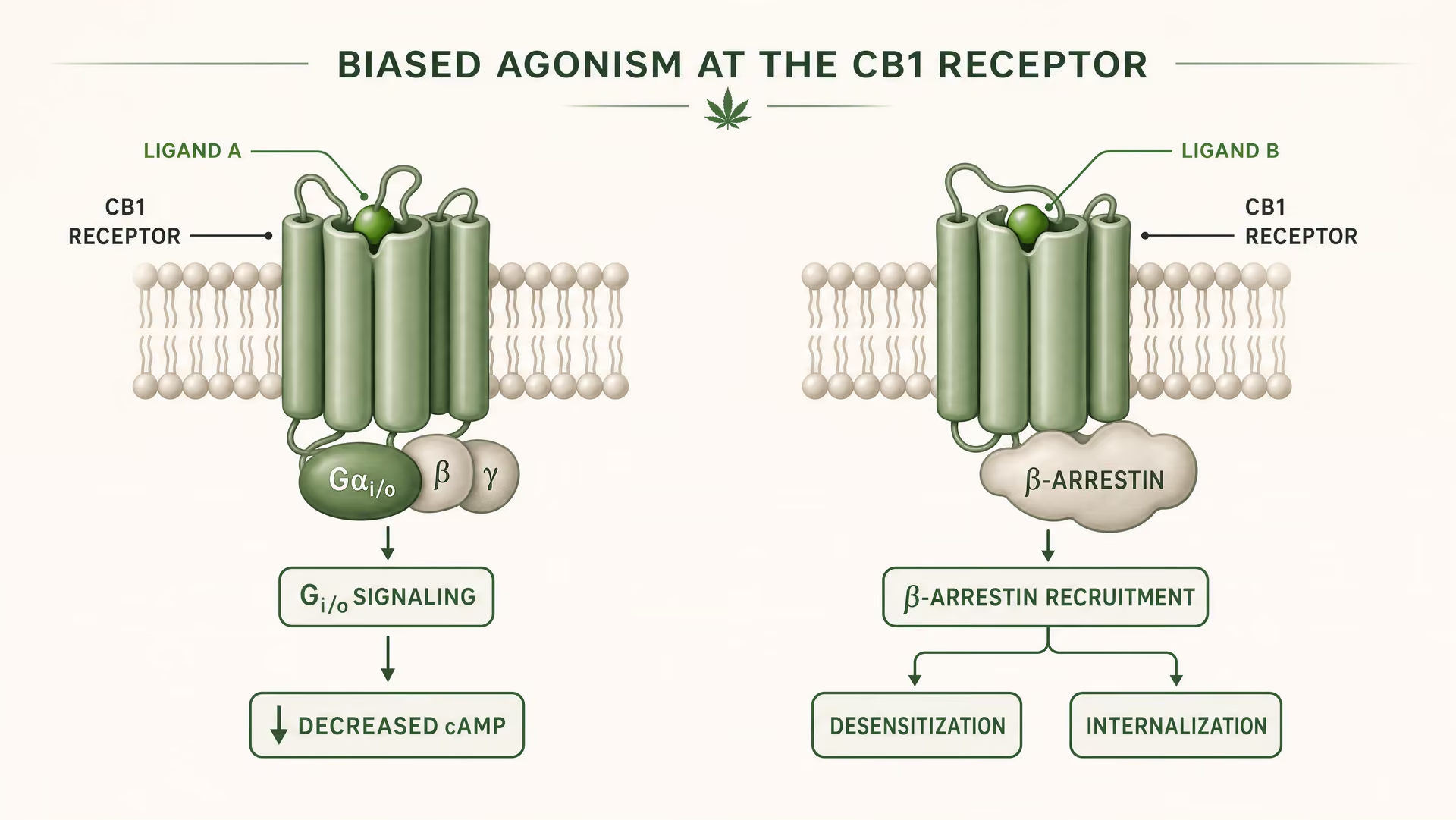
Was Verzerrter Agonismus In Der GPCR-Pharmakologie Bedeutet
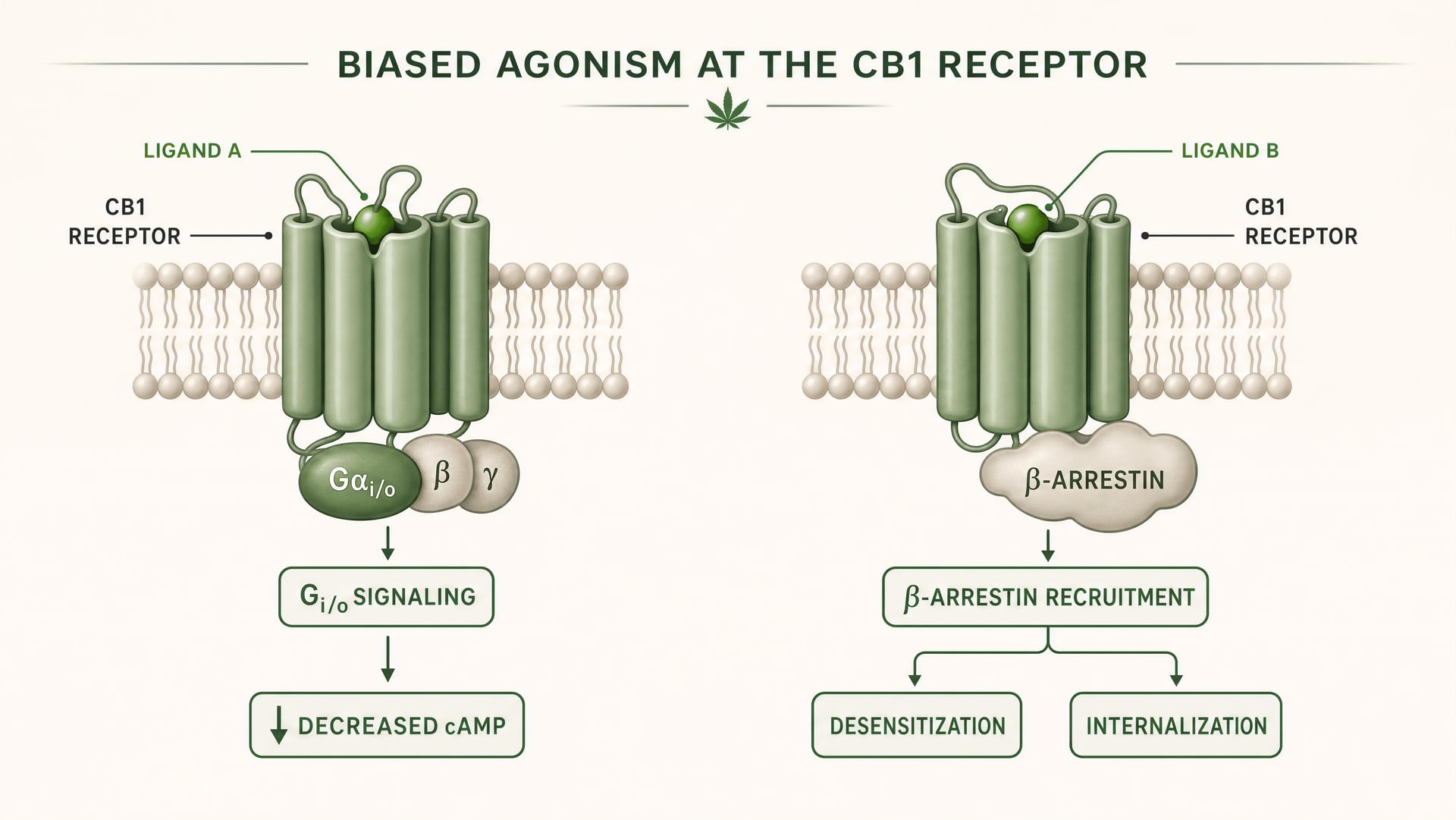
CB1 und CB2 sind G-Protein-gekoppelte Rezeptoren der Klasse A. Grundlagenarbeiten von Allyn Howlett etablierten CB1 als Gi/o-gekoppelten cannabinoid-Rezeptor und trugen dazu bei, das Feld von vager Pharmakologie hin zu rezeptordefinierten Mechanismen zu verschieben. Doch die Gi/o-Kopplung ist nur der Anfang der Geschichte. Sobald ein Ligand bindet, kann der Rezeptor mehr als eine aktive Form annehmen, und diese Formen signalisieren nicht identisch. Manche Rezeptorkonformationen begünstigen die Aktivierung von G-Proteinen. Andere rekrutieren beta-Arrestin stärker. Einige Zustände fördern die Phosphorylierung, Desensibilisierung oder Internalisierung des Rezeptors. Andere erzeugen ein länger anhaltendes Signaling von der Plasmamembran oder aus endosomalen Kompartimenten.
{kind=link}
Verzerrte Agonistik Eine Eigenschaft eines Liganden, der bei demselben Rezeptor bestimmte nachgeschaltete Signalwege gegenüber anderen bevorzugt.
Das ist biased agonism in einfachen Worten: Unterschiedliche Liganden stabilisieren unterschiedliche Rezeptorkonformationen, und diese Konformationen begünstigen unterschiedliche nachgeschaltete Signalwege. Ein Rezeptor ist nicht bloß an oder aus. Er ist konformationsgesteuert.
Für CB1 ist das besonders wichtig, weil der Rezeptor in einer Signallandschaft sitzt, die dicht, plastisch und stark zelltypspezifisch ist. In einer kortikalen glutamatergen Endigung kann ein Ligand die Transmitterfreisetzung über Gi/o-vermittelte Hemmung der Adenylylcyclase und Modulation von Ionenkanälen verringern. In einem GABAergen Interneuron kann derselbe Rezeptor die lokale Schaltkreisbalance in eine ganz andere Richtung verschieben. Wenn der Ligand außerdem eine starke beta-Arrestin-Rekrutierung fördert, kann der Rezeptor schneller internalisieren, wodurch ein Effekt verkürzt wird, während ein anderer eröffnet wird. Das Timing verändert sich. Der Ort des Signals verändert sich. Die physiologische Antwort verändert sich.
Das ist keine theoretische Haarspalterei. Die strukturelle Übersichtsarbeit von 2026 in Frontiers in Chemical Biology zu cannabinoid-Rezeptoren macht diesen Punkt klar: Die Liganden-Selektivität an CB1 und CB2 hängt von strukturellen Unterschieden auf Rezeptorebene ab, die Bindung, Signaling-Effektivität und Rezeptorregulation verändern. Das Schlüsselwort ist hier Regulation. Ein Ligand kann eine ähnliche Affinität haben, sich aber in Wirksamkeit, Arrestin-Rekrutierung, Verweilzeit oder Neigung zur Auslösung von Desensibilisierung unterscheiden. Die 2025/2026 in PubMed indexierte Studie zum dynamischen Mechanismus der Subtyp-Selektivität treibt denselben Gedanken weiter, indem sie argumentiert, dass Selektivität aus konformationellen Dynamiken entsteht und nicht aus einem statischen Schlüssel-Schloss-Modell. Endocannabinoide, Phytocannabinoide und synthetische Liganden sollten daher nicht in einen Topf geworfen werden. Anandamide, entdeckt von Raphael Mechoulam und Lumír Hanuš, verhält sich nicht wie delta-9-tetrahydrocannabinol, und keines von beiden verhält sich wie eine hochoptimierte synthetische Sonde.
Verzerrtes Signaling erklärt auch, warum allosterische Modulatoren so großes Interesse wecken. Ein allosterischer Ligand aktiviert CB1 möglicherweise nicht direkt so wie ein orthosterischer Agonist, kann aber die Signaling-Präferenzen des Rezeptors umformen, einen Signalweg verstärken und einen anderen dämpfen. Das eröffnet einen Weg zur Feinsteuerung. Zumindest prinzipiell.
CB1-Verzerrtes Signaling Als Forschungsrichtung Bei Schizophrenie
Der Artikel im American Journal of Psychiatry von 2025 liefert das bisher stärkste Argument dafür, dass CB1-biased signaling nicht nur ein Konzept der Pharmakologie ist, sondern eine plausible therapeutische Strategie bei Schizophrenie. Diese Argumentation verdient Aufmerksamkeit, weil die Schizophrenieforschung cannabinoids bislang meist über Epidemiologie, Risikobeziehungen oder grobe Warnungen vor Psychosen betrachtet hat. Der AJP-Artikel verschiebt den Rahmen. Er fragt, ob das Problem nicht „cannabis“ im Allgemeinen oder selbst „CB1-Aktivierung“ im Allgemeinen ist, sondern welche CB1-Signaling-Zustände in welchen Schaltkreisen und für welche Dauer aktiviert werden.
Das ist eine viel bessere Frage.
CB1 gehört zu den am häufigsten vorkommenden GPCRs im Gehirn, mit starker Expression in Kortex, Hippocampus, Basalganglien und Kleinhirn, doch die reine Häufigkeit erklärt klinische Effekte nicht. Schizophrenie umfasst dysregulierte Salienz, Kognition, Wahrnehmung und Netzwerkkopplung über kortikale und subkortikale Systeme hinweg. Ein Rezeptor, der geeignet ist, glutamaterge, GABAerge und dopaminerge schaltkreisbezogene Aktivität zu modulieren, ist daher per Design relevant. Der AJP-Artikel argumentiert, dass biased CB1-Liganden therapeutisch nützliche Schaltkreiseffekte von Nachteilen wie kognitiver Beeinträchtigung, Angst, Dysphorie oder psychotomimetischen Reaktionen trennen könnten.
Das ist eine ehrgeizige Behauptung, aber keine spekulative Nebelkerze. Sie folgt dem breiteren GPCR-Feld, in dem pathwayspezifische Verzerrung die Sichtweise auf Opioid-, Angiotensin- und Dopaminrezeptor-Arzneimittel bereits verändert hat. Die translationale Hoffnung bei CB1 besteht darin, dass bestimmte Signaloutputs die kortikale Netzwerkfunktion verbessern oder abnorme Schaltkreiszustände abschwächen können, ohne das vollständige Nebenwirkungsprofil einer hochwirksamen CB1-Agonisierung zu reproduzieren.
Schizophrenie ist ein guter Prüfstein, weil die klinische Hürde hoch ist. Ein Kandidatenarzneimittel darf nicht nur das Verhalten in einem Nagetierassay verändern. Es muss vermeiden, Psychose, Sedierung und kognitive Störungen bei Menschen zu verschlimmern, die bereits für solche Probleme anfällig sind. Das macht Pfadselektivität unmittelbar mehr als nur eine Frage der medizinischen Chemie. Sie wird zu einer Sicherheitsanforderung.
Der AJP-Rahmen hilft auch, eine häufige Vereinfachung in Diskussionen über cannabis zu korrigieren. Delta-9-THC ist ein Phytocannabinoid mit partieller Agonistenaktivität an CB1, aber seine Effekte spiegeln Dosis, Zeitpunkt, Rezeptorreserve, lokalen endocannabinoid-Tonus und die Beteiligung verschiedener neuronaler Populationen wider. Ein synthetischer CB1-Ligand, der so konstruiert ist, dass er einen intrazellulären Weg bevorzugt, könnte ganz anders wirken als THC, selbst wenn beide „CB1 treffen“. Umgekehrt gilt dasselbe: Zwei Verbindungen, die beide in einem präklinischen, mit Schizophrenie assoziierten Endpunkt verbessern, könnten sich hinsichtlich Kognition oder Affekt deutlich unterscheiden, wenn eine arrestinlastige Signalgebung antreibt und die andere nicht. Die Rezeptoridentität allein kann den gesamten Phänotyp nicht vorhersagen.
Warum Pfadselektivität Für Sicherheit Und Wirksamkeit Wichtig Ist
Pfadselektivität ist wichtig, weil Wirksamkeit keine eindimensionale Größe ist. Ein cannabinoid-Arzneimittel kann potent und dennoch klinisch schlecht sein. Es kann selektiv für CB1 sein und trotzdem versagen. Es kann CB2 vollständig meiden und dennoch über Netzwerk-Crosstalk unerwünschte immunologische oder metabolische Effekte hervorrufen. Die in PubMed indexierte integrative Netzwerk-Analyse von 2025/2026 identifizierte CB1 und CB2 als hoch einflussreiche Knoten im endocannabinoid-System und ordnete ihr Signaling Stoffwechselwegen zu. Diese Systemperspektive ist wesentlich. Rezeptoren arbeiten nicht isoliert, und Pfadverzerrung an einem Knoten kann sich auf breitere physiologische Programme auswirken.
Für CB1 sind Sicherheitsbedenken offensichtlich. Eine starke zentrale CB1-Aktivierung kann Gedächtnisbeeinträchtigung, veränderte Wahrnehmung, Angst, Tachykardie und bei anfälligen Personen psychosebezogene Effekte auslösen. Jedes therapeutische Programm gegen Schmerz, Appetit, Stimmung, Abhängigkeit oder Schizophrenie muss dieses Risikoprofil berücksichtigen. Ein Ligand, der einen gewünschten Gi/o-vermittelten synaptischen Effekt erhält, während er beta-Arrestin-vermittelte Desensibilisierung oder andere unerwünschte Signalkaskaden begrenzt, könnte theoretisch das therapeutische Fenster erweitern. Aber „theoretisch“ ist entscheidend. Viele biased-Ligand-Programme in der GPCR-Pharmakologie haben gezeigt, dass Bias, der in einem Assaysystem gemessen wird, nicht immer in vivo vorhersagt. Zellhintergrund, Rezeptordichte, Effektor-Expression und Kinetik können die scheinbare Verzerrung alle verschieben.
CB2 bietet eine mahnende Parallele. Die Übersichtsarbeit von 2026 in Frontiers in Behavioral Neuroscience beschreibt ein Update der letzten 3 Jahre, in dem CB2-Signaling durch Verbindungen zu neuroinflammatorischen und neurodegenerativen Mechanismen im Zusammenhang mit Erkrankungen des zentralen Nervensystems Aufmerksamkeit erhielt. Das untergräbt direkt die alte Vorstellung, dass CB2 für das Gehirn irrelevant sei. Dennoch garantiert die bloße Zielsetzung auf CB2 kein nützliches entzündungshemmendes Arzneimittel. Die Verteilung ist gradueller als die alte Trennung zwischen Gehirn und Körper, und die Signaling-Folgen hängen weiterhin von Ligand und Kontext ab.
Die praktische Lehre ist daher klar: Rezeptor-Subtyp-Selektivität ist notwendig, aber nicht ausreichend. Pfadselektivität könnte den Unterschied ausmachen zwischen einem cannabinoid, das therapeutisch aussieht, einem, das intoxikierend ist, und einem, das in Studien versagt, weil es Nutzen und Nebenwirkung nicht trennen kann. Für CB1, besonders in der Psychiatrie, wird genau diese Unterscheidung wahrscheinlich entscheiden, ob der Rezeptor eine Warnungsgeschichte bleibt oder zu einem praktikablen Arzneimittelziel wird.
Strukturbiologie von CB1 und CB2: Wie Form Selektivität bestimmt
Die Strukturbiologie hat die Art verändert, wie über cannabinoid receptors gesprochen wird. Die alte Kurzformel — CB1 erklärt Intoxikation, CB2 erklärt Entzündung — übersieht die Tatsache, dass beide Rezeptoren Klasse-A-G-Protein-gekoppelte Rezeptoren sind, deren Verhalten von Form, Bewegung, Bindungstiefe und den in einer bestimmten Zelle verfügbaren Signalpartnern abhängt. Das ist weit über die Grundlagenpharmakologie hinaus bedeutsam. Nach Angaben der WHO verwendeten 2019 schätzungsweise 200 Millionen Menschen cannabis, also 4 % der Weltbevölkerung im Alter von 15 bis 64 Jahren, doch die FDA listet im Jahr 2025 weiterhin nur ein cannabis-abgeleitetes Arzneimittel und drei cannabis-bezogene Arzneimittelprodukte als zugelassen. Ein Grund für diese Lücke ist struktureller Natur: Es ist schwierig, cannabinoid Liganden zu entwickeln, die den richtigen Rezeptor, auf die richtige Weise und für die richtige Dauer ansprechen.
Die 2026 in Frontiers in Chemical Biology veröffentlichte Übersichtsarbeit macht diesen Punkt deutlich. CB1 und CB2 unterscheiden sich nicht nur darin, wo sie exprimiert werden. Sie unterscheiden sich in der Architektur ihrer Ligandenbindungs-Taschen, der Form und Flexibilität ihrer extrazellulären Schleifen, der Packung ihrer Transmembranhelices und den Konformationszuständen, die sie nach Ligandenbindung bevorzugen. Diese Eigenschaften beeinflussen nicht nur die Selektivität, sondern auch Wirksamkeit, Desensibilisierung, Internalisierung und Pathway Bias.
Was Strukturstudien über Rezeptortaschen offenbaren
Eine orthosterische Tasche ist die Haupt-Bindungstasche, in der endogene Liganden wie Anandamid und 2-Arachidonoylglycerol, Phytocannabinoide wie THC und viele synthetische Liganden ihren primären Kontakt eingehen. Bei CB1 und CB2 liegt diese Tasche innerhalb des Bündels aus sieben Transmembranhelices, teilweise abgeschlossen durch extrazelluläre Schleifen, die den Zugang entweder öffnen oder einschränken können.
Cryo-EM- und Röntgenstrukturen der letzten Jahre zeigten, dass cannabinoid Rezeptoren sich nicht wie starre Schlösser verhalten, die auf einen Schlüssel warten. Sie lassen sich besser als bewegliche Zielstrukturen mit bevorzugten Formen verstehen. Die Frontiers-in-Chemical-Biology-Übersichtsarbeit von 2026 betont, dass die orthosterischen Hohlräume von CB1 und CB2 sich in ihrer Fähigkeit, überlappende Ligandenklassen zu binden, ähneln, sich jedoch in Größe, Restidentität und lokaler Flexibilität so weit unterscheiden, dass Affinität und Signalantwort verschoben werden. Deshalb können eng verwandte Verbindungen pharmakologisch auseinanderfallen. Eine kleine Veränderung in Substituentengröße, Polarität oder Schwanzlänge kann verändern, wie tief ein Ligand in die Tasche eindringt, welche Helices er verschiebt und ob der Rezeptor in einen G-Protein-bevorzugenden oder Arrestin-bevorzugenden Zustand übergeht.
CB1 war strukturell besonders aufschlussreich, weil inzwischen viele hochaufgelöste inaktive und aktive Modelle existieren. Ein wiederkehrendes Thema ist, dass seine Tasche weitläufig und hydrophob ist und damit der lipophilen Natur vieler cannabinoids entspricht. Extrazelluläre Schleife 2 und die oberen Abschnitte mehrerer Helices formen den Zugangskanal. Transmembranhelices sind die sieben membranüberspannenden Segmente, aus denen der Rezeptorkern besteht; wenn ein Ligand bindet, können sich diese Helices relativ zueinander verschieben. Die pharmakologisch wichtigste Bewegung findet meist auf der intrazellulären Seite statt, wo eine nach außen gerichtete Bewegung von Helix 6 eine Andockstelle für Gi/o-Proteine schafft. Diese Verschiebung ist eines der Kennzeichen der Rezeptoraktivierung.
CB2 teilt denselben grundlegenden GPCR-Faltungstyp, doch die Frontiers-Übersichtsarbeit argumentiert, dass subtypspezifische Aminosäureunterschiede rund um die Tasche und die Schleifenregionen der Wirkstoffchemie nutzbare Hebel für Selektivität bieten. Es geht dabei nicht einfach darum, dass eine Tasche „hirnähnlich“ und die andere „immunähnlich“ sei. Der Punkt ist geometrischer und energetischer Natur. Unterschiedliche Reste verändern die Kontur der Kavität, lokale Wasserstoffbrückenoptionen, aromatisches Stapeln und die Flexibilität von Zugangskanälen, durch die Liganden aus der Membran eintreten.
Eine 2025/2026 in PubMed indexierte Studie zum dynamischen Mechanismus der Subtyp-Selektivität ging noch weiter und argumentierte, dass die endocannabinoid Selektivität nicht nur eine Frage statischer Bindungsaffinität ist. Konformationsdynamik ist entscheidend. Einfach ausgedrückt kann ein Rezeptor vor und nach Ligandenbindung mehrere Formen durchlaufen, und manche Liganden stabilisieren eine selektive Form besser als andere. Das hilft zu erklären, warum endogene Lipide, Phytocannabinoide und synthetische Liganden unterschiedliche Subtyppräferenzen zeigen können, selbst wenn ihre Gerüste auf dem Papier verwandt aussehen.
Determinanten der Ligandenselektivität zwischen CB1 und CB2
Selektivität beginnt mit der Kontaktchemie, endet dort aber nicht. Die Frontiers-in-Chemical-Biology-Übersichtsarbeit beschreibt Selektivität als Produkt struktureller Unterschiede auf Rezeptorebene, die Bindung, Signalwirksamkeit und Regulation zugleich beeinflussen. Das ist die richtige Perspektive. Ein Ligand kann in einem Radioliganden-Bindungsassay CB2-selektiv sein, diesen praktischen Vorteil aber verlieren, wenn er zugleich Rezeptorzustände fördert, die in krankheitsrelevanten Zellen zu rascher Toleranz oder schwacher Signalgebung führen.
Mehrere strukturelle Merkmale tauchen immer wieder auf. Erstens unterscheidet sich die Aminosäurezusammensetzung der orthosterischen Tasche zwischen CB1 und CB2 in einem Maß, das verändert, wie Kopfgruppe, Grundgerüst und hydrophober Schwanz eines Liganden aufgenommen werden. Zweitens helfen extrazelluläre Schleifen, den Eintritt und die Orientierung zu formen. Drittens können die oberen und mittleren Bereiche der Transmembranhelices den Rezeptor in leicht unterschiedliche aktive Zustandsensembles verschieben. Ein Konformationszustand ist einfach eine der möglichen Formen des Rezeptors in einem gegebenen Moment. Verschiedene Liganden binden nicht nur an einen Rezeptor; sie stabilisieren eine Teilmenge dieser Formen.
Deshalb ist die Subtyp-Selektivität bei natürlichen cannabinoids oft nur mäßig. THC etwa interagiert mit beiden Rezeptoren. Anandamid und 2-AG wirken ebenfalls an beiden, wenn auch mit kontextabhängigen Unterschieden in Potenz, Wirksamkeit und Metabolismus. Synthetische Liganden waren nützlicher, um Struktur-Selektivitäts-Beziehungen zu entwirren, weil Chemiker Merkmale wie Seitenkettenlänge, Ringspannung und polare Substituenten systematisch verändern können. Selbst dann bleibt eine saubere Trennung schwierig. CB1 und CB2 sind so homolog, dass eine für den einen entwickelte Verbindung häufig weiterhin eine relevante Aktivität am anderen zeigt.
Das hat praktische Konsequenzen. Arzneimittelentwickler strebten lange CB2-selektive Agonisten an, in der Hoffnung, antiinflammatorische oder analgetische Effekte ohne starke CB1-vermittelte zentrale Nebenwirkungen zu erzielen. Manchmal funktioniert diese Strategie pharmakologisch, aber sie ist kein Freifahrtschein. Die 2026 in Frontiers in Behavioral Neuroscience erschienene Übersichtsarbeit betont, dass CB2 in den letzten 3 Jahren verstärkt bei Störungen des zentralen Nervensystems in den Fokus geraten ist, was die vereinfachte Sicht untergräbt, CB2 sei für das Gehirn irrelevant. Selbst ein „peripherer“ CB2-Ligand kann daher nicht mit veralteten Rezeptorkarten beurteilt werden.
Warum Wirksamkeit und Regulation ebenfalls strukturelle Fragen sind
Potenz beantwortet, wie viel Ligand benötigt wird. Wirksamkeit fragt, was dieser Ligand nach der Bindung mit dem Rezeptor bewirkt. Die Strukturbiologie verbindet beides, vermischt es aber nicht. Zwei Liganden können dieselbe Tasche besetzen und sehr unterschiedliche Antworten auslösen, weil sie unterschiedliche aktive Konformationen stabilisieren.
Bei CB1 ist das zentral für das heutige therapeutische Denken. Der 2025 im American Journal of Psychiatry erschienene Artikel argumentiert, dass biased signaling an CB1 eine plausible Strategie für Schizophrenie sein könnte, eine Erkrankung, von der weltweit etwa 24 Millionen Menschen betroffen sind. Biased Agonism bedeutet, dass ein Ligand einen Signalweg gegenüber einem anderen bevorzugt, häufig Gi/o-Signalgebung gegenüber beta-arrestin recruitment oder umgekehrt. Strukturell entsteht dieser Bias dadurch, dass der Ligand die intrazelluläre Seite des Rezeptors in eine Form verschiebt, die besser zu einem Signalpartner als zu einem anderen passt. Das ist keine abstrakte Idee. Es ist ein Ziel der Wirkstoffchemie.
Beta-Arrestins sind wichtig, weil sie zu Desensibilisierung und Internalisierung beitragen. Desensibilisierung bedeutet, dass der Rezeptor nach wiederholter oder anhaltender Aktivierung weniger reagiert. Internalisierung bedeutet, dass der Rezeptor von der Zelloberfläche in das Zellinnere aufgenommen wird. Beide Prozesse werden von der Rezeptorkonformation beeinflusst. Manche Liganden aktivieren G-Proteine stark, rekrutieren Arrestine aber nur schwach; andere tun beides. Ein behaviorally tolerierbares CB1-Arzneimittel könnte genau diese Art der Trennung benötigen, statt einfacher Blockade oder Vollagonismus.
Allosterische Modulation fügt eine weitere Ebene hinzu. Ein allosterischer Ligand bindet außerhalb der orthosterischen Tasche und verändert, wie der Rezeptor auf andere Liganden reagiert. Strukturell ist das attraktiv, weil es eine feinere Kontrolle der Rezeptorform ermöglichen könnte als das starke Besetzen der Haupttasche mit einem hochwirksamen Agonisten. Für die cannabinoid Pharmakologie könnte das bedeuten, nützliche Signalgebung zu erhalten und gleichzeitig unerwünschte Wirkungen zu reduzieren, die mit einer übermäßigen CB1-Aktivierung verbunden sind.
Die größere Lehre ist eindeutig. Rezeptorform ist kein Nebendetail. Sie erklärt, warum Phytocannabinoide, endocannabinoids und synthetische Verbindungen unterschiedliche klinische Profile hervorrufen können, selbst wenn sie auf dieselbe Rezeptorfamilie abzielen. Da CB1 und CB2, wie eine 2025/2026 in PubMed indexierte Systemstudie berichtete, an einflussreichen Knotenpunkten in Netzwerkanalysen des endocannabinoid systems sitzen, können Fehler bei Selektivität oder Wirksamkeit sich über viele Signalwege hinweg fortpflanzen. Strukturelle Einsicht garantiert kein erfolgreiches Arzneimittel. Sie erklärt jedoch, warum es so schwierig bleibt, Selektivität, Potenz und Verträglichkeit miteinander in Einklang zu bringen.
Endocannabinoids und Subtypenspezifität: Anandamid, 2-AG und dynamisches Rezeptorverhalten
Die Pharmacologie von endocannabinoids wird oft mit einer Abkürzung erklärt, die ordentlich klingt, aber in die Irre führt: Anandamid und 2-Arachidonoylglycerol (2-AG) werden behandelt, als wären sie die eingebauten „Standardagonisten“ des Körpers, nützlich vor allem als Referenzpunkte dafür, was CB1 und CB2 normalerweise tun. Diese Darstellung verfehlt den zentralen Befund. Es handelt sich nicht um stabile, frei zirkulierende Hormone, die Rezeptoren unter einheitlichen Bedingungen begegnen. Sie sind kurzlebige Lipidmediatoren, die bei Bedarf in Membranen durch zelltypspezifische Enzymsysteme gebildet und dann rasch abgebaut werden. Ihre scheinbare Rezeptorpräferenz kann sich mit der Membranzusammensetzung, den Rezeptorkonformationen, der lokalen Enzymaktivität und dem Zeitpunkt der Freisetzung verschieben.
Das ist nicht nur für die akademische Rezeptortheorie bedeutsam. Cannabis wurde 2019 schätzungsweise von 200 Millionen Menschen konsumiert, also von 4 % der Weltbevölkerung im Alter von 15 bis 64 Jahren, laut WHO, und auf cannabinoidzielende Arzneimittel wird bereits klinisch zurückgegriffen: Die FDA erklärte 2025, dass sie ein aus cannabis gewonnenes Arzneimittel und drei cannabisbezogene Arzneimittel zugelassen habe. Wenn endogene Liganden sich nicht wie einfache Lehrbuch-Kontrollen verhalten, dann ist der Vergleich von Phytocannabinoids oder Arzneimittelkandidaten mit ihnen nicht unkompliziert.
Warum endogene Liganden keine einfachen Referenzverbindungen sind
Anandamid, 1992 von Devane, Hanus, Breuer, Mechoulam und Kolleginnen und Kollegen identifiziert, und 2-AG, wenige Jahre später als weiterer wichtiger endocannabinoid etabliert, unterscheiden sich in mehr als nur Potenztabellen. Sie entstehen über unterschiedliche Biosynthesewege, erreichen Rezeptoren in unterschiedlichen Zeitskalen und werden durch unterschiedliche Enzyme entfernt. Anandamid wird vor allem aus N-Arachidonoylphosphatidylethanolamin über Wege gebildet, an denen NAPE-PLD beteiligt ist, obwohl alternative Wege existieren. 2-AG wird überwiegend aus Diacylglycerol durch DAGLα und DAGLβ produziert. Das sind keine nebensächlichen biochemischen Fußnoten; sie bestimmen, wo und wann Rezeptoraktivierung stattfindet.
Selbst die ältere Aussage, Anandamid sei ein CB1-präferierender Ligand, während 2-AG ein Vollagonist an CB1 und CB2 sei, kann zu grob werden, wenn man sie von gereinigten Systemen auf lebendes Gewebe überträgt. Anandamid ist außerdem ein Substrat der fatty acid amide hydrolase, oder FAAH, und kann unter bestimmten Bedingungen nicht-cannabinoidale Ziele wie TRPV1 ansprechen. 2-AG liegt in Hirngewebe in vielen Messungen in deutlich höheren basalen Konzentrationen vor als Anandamid und wird hauptsächlich durch monoacylglycerol lipase, oder MAGL, hydrolysiert, wobei auch ABHD6 und ABHD12 beitragen. Das „Input“-Signal, das jeder Ligand liefert, ist also nicht nur eine Frage von Affinität oder Wirksamkeit an CB1 versus CB2. Entscheidend ist auch, ob der Ligand an der Synapse, in einer Immunzelle, nahe einer intrazellulären Membran oder in einem Kompartiment mit vielen abbauenden Enzymen gebildet wird.
Die Arbeiten von Allyn Howlett zur Rezeptorpharmacologie halfen dabei, CB1 als bona fide GPCR zu etablieren, der überwiegend an Gi/o-Proteine gekoppelt ist. Dieses Rahmenmodell bleibt wesentlich, stützt aber kein einfaches Modell von endogenem Referenzarzneimittel. CB1 und CB2 können über Hemmung der Adenylylcyclase, Regulation von Ionenkanälen, MAP-Kinase-Wege, Rekrutierung von beta-arrestin, Rezeptorinternalisierung und Desensibilisierung signalisieren. Ein Ligand, der in einem Assay „schwächer“ wirkt, kann in einem Gewebe mit langsamer Degradation eine längere Signalwirkung erzeugen oder einen Signalweg gegenüber einem anderen bevorzugen. Der 2025 erschienene Artikel im American Journal of Psychiatry über CB1-biased signaling macht diesen Punkt in einem Krankheitskontext deutlich: Die therapeutische Frage bei Störungen wie Schizophrenia, von der weltweit etwa 24 Millionen Menschen betroffen sind, lautet nicht nur, ob CB1 aktiviert wird, sondern wie.
Dynamische Mechanismen der Subtypenselektivität
Die kürzlich in PubMed indexierte Studie zur Subtypenselektivität (PMID: 41962866) ist wichtig, weil sie das Feld von einer Schloss-und-Schlüssel-Erzählung wegführt. Ihre zentrale Botschaft ist, dass endocannabinoid Selektivität aus dynamischen Mechanismen entstehen kann. Mit anderen Worten: Die Präferenz für einen Rezeptorsubtyp lässt sich nicht allein durch einen festen Unterschied darin erklären, wie fest ein Ligand CB1 versus CB2 bindet. Der Ligand-Rezeptor-Komplex durchläuft mehrere Konformationszustände, und diese Zustände unterscheiden sich zwischen CB1 und CB2.
Dieser Befund passt zu der 2026 in Frontiers in Chemical Biology erschienenen Übersichtsarbeit, die argumentiert, dass strukturelle Unterschiede zwischen CB1 und CB2 Bindung, Signalwirksamkeit und Rezeptorregulation prägen. Die Rezeptoren teilen eine erhebliche Sequenzähnlichkeit, doch kleine Unterschiede in der Architektur der orthosterischen Tasche, den extrazellulären Schleifen, den Bewegungen der Transmembranhelices und den intrazellulären Kopplungsflächen können beeinflussen, was nach der Ligandenbindung geschieht. Ein Ligand kann in beide Rezeptoren ähnliche Taschen einnehmen, aber in einem Subtyp einen stärker signalaktiven Zustand stabilisieren oder unterschiedliche Übergänge zwischen inaktiven, intermediären und aktiven Zuständen begünstigen.
Für endocannabinoids ist das besonders plausibel, weil es sich um flexible Lipide und nicht um starre synthetische Gerüste handelt. Flexibilität ist eine pharmakologische Variable. Anandamid kann mehrere Konformationen annehmen, und 2-AG ist in Membranen ebenfalls dynamisch. Die Frage lautet dann nicht nur: „Bindet es?“, sondern: „Welche Rezeptorzustände begünstigt es, wie lange werden diese Zustände besetzt, und welche Proteine stehen als Nächstes zur Kopplung zur Verfügung?“ Ein CB1-reiches Neuron und eine CB2-exprimierende Mikrogliazelle bieten nicht dasselbe Signalmilieu, selbst bevor Unterschiede in der Rezeptordichte berücksichtigt werden.
Diese dynamische Sicht schwächt auch die alte Annahme, dass endogene Liganden eine einzige physiologische Baseline definieren, an der THC, cannabidiol oder synthetische cannabinoids gemessen werden können. THC ist ein phytocannabinoid mit eigenem Wirksamkeitsmuster und kinetischem Verhalten. Endocannabinoids sind ereignisgesteuerte Signale, oft phasisch und lokal. Ein synthetischer CB2-Agonist kann in einem rekombinanten Assay selektiv erscheinen, aber in Gewebe unerwartete Ergebnisse erzeugen, wenn Rezeptorreserve, Membran-Cholesterin, Heteromer-Bildung oder beta-arrestin-Verarbeitung anders ausfallen. Selektivität auf dem Papier ist nicht das Schicksal.
Wie lokale Synthese und Degradation die Rezeptorbindung prägen
In vivo wird die endocannabinoid Signalübertragung ebenso sehr durch Enzymologie wie durch Rezeptorpharmacologie bestimmt. An vielen zentralen Synapsen wird 2-AG postsynaptisch als Antwort auf Calcium-Anstiege oder Gq/11-gekoppelte Rezeptoraktivierung gebildet, wandert dann retrograd zu präsynaptischen CB1-Rezeptoren und unterdrückt die Neurotransmitterfreisetzung. Das ist ein zeitliches Steuerungssystem im Bereich von Sekunden, kein diffuser Hintergrundton. Das Signal endet, wenn MAGL und verwandte Hydrolasen 2-AG entfernen. Anandamid verhält sich oft anders, mit geringerer Gewebeabundanz, anderen Freisetzungsbedingungen und stärkerer Empfindlichkeit gegenüber dem FAAH-gesteuerten Abbau.
Das bedeutet: Eine Veränderung eines Enzyms kann die Rezeptorbindung verändern, ohne den Rezeptor selbst zu verändern. Eine FAAH-Hemmung erhöht den Anandamid-Tonus, aber das physiologische Ergebnis hängt davon ab, wo Anandamid gebildet wird, ob TRPV1 ebenfalls vorhanden ist und ob CB1-Rezeptoren in diesem Mikrodomain desensibilisiert oder internalisiert sind. Eine MAGL-Hemmung kann 2-AG deutlich erhöhen, aber eine chronische Erhöhung kann in manchen Systemen zu CB1-Desensibilisierung führen. Mehr Ligand bedeutet nicht immer mehr nützliches Signal.
Die systembiologische Studie mit PMID: 42129940 stützt dieses breitere Bild, indem sie CB1 und CB2 als einflussreiche Knoten in Verbindung mit Stoffwechselwegen und nicht als isolierte Schalter einordnet. Das ist die richtige Ebene, um endocannabinoid Wirkung zu verstehen. Ligandenverfügbarkeit, Membranzugang, Rezeptorkonformation, G-Protein-Kopplung, beta-arrestin-Rekrutierung und Degradation greifen ineinander. Ebenso tun es Gewebegradienten. CB2 ist nicht einfach „außerhalb des Gehirns“, und CB1 ist nicht nur „der Rezeptor für Psychoaktivität“. Die endocannabinoid Signalübertragung ist lokal, bedingt und kinetisch. Jeder Bericht über Subtypenselektivität, der diese Merkmale ignoriert, wird sowohl Physiologie als auch Arzneimittelentwicklung falsch lesen.
Das Endocannabinoid-System als Netzwerk, nicht als Zwei-Rezeptor-Diagramm
Das Endocannabinoid-System ergibt wenig Sinn, wenn man es als zwei Kreise mit den Bezeichnungen CB1 und CB2 zeichnet, von denen Pfeile nach außen zeigen. Diese schematische Darstellung war in der Frühphase des Fachgebiets hilfreich, insbesondere nach Allyn Howletts Arbeiten zur Rezeptorpharmakologie und der Identifizierung von Anandamid durch Raphael Mechoulam und Lumír Hanuš, verdeckt heute jedoch mehr, als sie erklärt. Die praktischen Konsequenzen sind erheblich. Die World Health Organization schätzte, dass im Jahr 2019 200 Millionen Menschen cannabis konsumierten, etwa 4 % der globalen Bevölkerung im Alter von 15 bis 64 Jahren, während die U.S. FDA angibt, dass bis 2025 ein aus cannabis abgeleitetes Arzneimittel und drei cannabisbezogene Arzneimittel zugelassen sind. Ein Rezeptorsystem, das so viele Expositionen und so viele therapeutische Ansprüche berührt, kann nicht als schlichtes Paar aus Hirnrezeptor und Immunrezeptor behandelt werden.
Eine systemische Sichtweise geht von einer grundlegenden Tatsache aus: Endocannabinoide sind Lipide, die nach Bedarf gebildet, rasch abgebaut und in denselben biochemischen Raum eingebettet werden wie Entzündungsmediatoren, Wege des Membran-Remodelings und synaptische Rückkopplungsschleifen. Anandamid und 2-Arachidonoylglycerol sind keine isolierten „Botschaften“. Sie werden aus Membranvorstufen gebildet, konkurrieren mit anderen Lipidwegen um Substrate und wirken in Geweben, in denen Rezeptordichte, Zelltyp, Enzymexpression und Signalpartner jeweils unterschiedlich sind. Deshalb kann derselbe Ligand in einer Situation anxiolytisch wirken, in einer anderen intoxizierend, an anderer Stelle antiinflammatorisch und in einer klinischen Studie enttäuschend bleiben.
Was die integrative Netzwerkanalyse hinzufügt
Die in PubMed indexierte integrative Netzwerkanalyse aus dem Jahr 2025/2026 (PMID: 42129940) ist nützlich, weil sie die Aufmerksamkeit von Einzelzielen auf die Systemorganisation verschiebt. Statt nur zu fragen, welcher Ligand an CB1 oder CB2 bindet, kartieren die Autoren das Endocannabinoid-System als Interaktionsnetzwerk, das Rezeptoren, biosynthetische und degradative Enzyme, lipidische Zwischenprodukte sowie Verbindungen auf Pathway-Ebene zu Entzündung, Stoffwechsel und neuronaler Signalübertragung umfasst. In diesem Rahmen wird Einfluss nicht allein durch die Rezeptorhäufigkeit definiert. Er wird durch Konnektivität und dadurch bestimmt, wie sich Störungen durch das Netzwerk ausbreiten.
Das ist für die Interpretation wichtig. Wenn ein Arzneimittel die CB1-Signalgebung verändert, ist die nachgeschaltete Wirkung nicht auf eine Gi/o-vermittelte Hemmung der Adenylylcyclase beschränkt. Sie kann die Neurotransmitterfreisetzung verändern, das Verhalten von Calcium- und Kaliumkanälen verschieben, die Rekrutierung von Beta-Arrestin verändern, die Rezeptor-Internalisierung auslösen und indirekt den Fluss von Lipidsubstraten modifizieren, die auch eicosanoid- und andere entzündungsbezogene Wege speisen. Ein Netzwerkmodell erfasst diese systemübergreifenden Konsequenzen besser als ein Diagramm zur Rezeptorbelegung.
Hier zeigt sich auch, warum die alte Kurzformel „CB1 im Gehirn, CB2 in Immunzellen“ unzureichend ist. Die Verteilung ist abgestuft, zelltypspezifisch und zustandsabhängig. CB1 ist in vielen neuronalen Populationen reichlich vorhanden, jedoch nicht gleichmäßig im gesamten Gehirn, und es kommt auch in peripheren Geweben vor, die für Energiehaushalt, Nozizeption und Organfunktion relevant sind. CB2 ist in Immunzelllinien angereichert, doch neuere Arbeiten zum zentralen Nervensystem haben die Vorstellung, es sei für die Hirnbiologie irrelevant, deutlich hinter sich gelassen. Eine 2026 in Frontiers in Behavioral Neuroscience veröffentlichte Übersichtsarbeit beschrieb dies als „ein Update der letzten 3 Jahre“ und betonte CB2-Signalgebung in neuroinflammatorischen und neurodegenerativen Kontexten. Entscheidend ist nicht, dass CB2 plötzlich „auch der Hirnrezeptor“ wurde. Entscheidend ist, dass die Bedeutung eines Rezeptors davon abhängt, welche Zellen aktiv, verletzt, entzündet oder adaptiv verändert sind.
Die Netzwerkanalyse hilft zu erklären, warum rezeptororientierte Arzneimittel oft breitere Effekte haben als erwartet. Ein CB1-aktives Phytocannabinoid wie Delta-9-THC, ein Endocannabinoid wie Anandamid und ein synthetischer Agonist können denselben Rezeptor aktivieren und dennoch unterschiedliche zeitliche und räumliche Verläufe auslösen. Die Ligandenklasse ist wichtig. Ebenso Wirksamkeit, Verweildauer, Metabolismus und der Zugang zu unterschiedlichen Rezeptorkonformationen.
CB1 und CB2 als einflussreiche Knoten
Die integrative Netzwerkstudie identifiziert CB1 und CB2 als hoch einflussreiche Knoten, nicht weil sie isoliert arbeiten, sondern weil sie an Kontrollpunkten liegen, an denen viele Wege zusammenlaufen. In Netzwerkbegriffen sind einflussreiche Knoten Orte, an denen lokale Veränderungen sich weit ausbreiten können. In pharmakologischen Begriffen sind es Rezeptoren, deren Aktivierungszustand synaptische Signalgebung, Immunreaktionen und zelluläre Stressprogramme gleichzeitig umleiten kann.
CB1 ist das klarere Beispiel dafür, warum „einflussreich“ nicht „einfach“ bedeutet. Es signalisiert überwiegend über Gi/o-Proteine, doch das ist nur der Anfang. Abhängig von Ligand und Kontext kann CB1 Beta-Arrestine rekrutieren, eine phosphorylierungsabhängige Desensibilisierung durchlaufen, internalisieren, recyceln oder an der Membran dauerhaft aktiv bleiben. Der Artikel im American Journal of Psychiatry aus dem Jahr 2025 argumentiert, dass Bias-Signalgebung an CB1 eine plausible therapeutische Strategie für Schizophrenie sei, eine Erkrankung, von der die WHO angibt, dass sie weltweit etwa 24 Millionen Menschen betrifft. Dieser Vorschlag ergibt nur dann Sinn, wenn CB1 als Signal-Hub mit trennbaren Ausgängen verstanden wird, nicht als Ein/Aus-Schalter für Intoxikation. Ein Ligand, der einen Signalzweig gegenüber einem anderen begünstigt, kann einige therapeutische Effekte erhalten und unerwünschte Effekte verringern. Kann, nicht wird. Bias kann Belastungen reduzieren, hebt aber nicht auf, dass CB1 in Schaltkreisen liegt, die Kognition, Belohnung, motorische Kontrolle, Schmerzverarbeitung und Appetit regulieren.
CB2 zeigt ein ähnliches Problem aus der entgegengesetzten Richtung. Es wurde lange als das sicherere Ziel angesehen, weil es weniger mit akuten psychoaktiven Effekten verbunden war. Diese Sichtweise ist zu optimistisch. CB2-gerichtete Liganden können dennoch enttäuschende oder gemischte Ergebnisse hervorrufen, wenn krankheitsrelevante Signalgebung vom Zellzustand, von der Induzierbarkeit des Rezeptors, vom Entzündungstonus oder von der Kreuzkommunikation mit metabolischen Enzymen abhängt. Die aktuelle strukturelle Übersicht in Frontiers in Chemical Biology, veröffentlicht im Jahr 2026, betont, dass die Selektivität zwischen „CB1 und CB2“ von strukturellen Unterschieden auf Rezeptorebene abhängt, die Bindung, Wirksamkeit und Rezeptorregulation verändern. Die 2025/2026 in PubMed indexierte Studie zur Subtyp-Selektivität (PMID: 41962866) geht noch weiter und argumentiert, dass Selektivität dynamisch ist und durch konformationelles Verhalten geprägt wird, nicht durch eine statische Schloss-und-Schlüssel-Passung. Das ist ein starkes Argument dafür, nicht länger von CB1-selektiven oder CB2-selektiven Verbindungen zu sprechen, als wäre Selektivität eine feste Eigenschaft mit festen biologischen Konsequenzen.
Verbindungen zwischen Rezeptorsignalisierung und Stoffwechselwegen
Der wertvollste Beitrag der Netzwerkperspektive besteht darin, dass sie Rezeptorpharmakologie mit dem Lipidstoffwechsel wieder verknüpft. Die Endocannabinoid-Signalgebung ist auf einer Chemie aufgebaut, die mit Arachidonsäure zusammenhängt. Anandamid und 2-AG werden durch Enzymsysteme gebildet und abgebaut, die auch die Verfügbarkeit bioaktiver Lipide beeinflussen, die an Entzündung und Gewebereaktion beteiligt sind. Ein Arzneimittel, das FAAH oder MAGL hemmt, „erhöht“ nicht bloß Endocannabinoide. Es verteilt den Signalverkehr in einem metabolischen Netzwerk um.
Das kann nützlich sein. Es kann aber auch nach hinten losgehen. Die Erhöhung von 2-AG kann beispielsweise in einigen Kompartimenten die CB1- oder CB2-Signalgebung verstärken, während sie in anderen durch Substratumlagerung oder nachgeschaltete Oxidation prostaglandinbezogene Wege verändert. Die klinische Konsequenz ist eindeutig: Rezeptororientierte und enzymorientierte Strategien können systemweite Folgen haben, selbst wenn ihr Designziel eng gefasst ist. Das ist ein Grund dafür, dass selektive Verbindungen trotz hohen ungedeckten Bedarfs nicht automatisch zu klaren klinischen Profilen bei Schmerz, psychiatrischen Erkrankungen oder Entzündungsstörungen geführt haben. Allein chronische Schmerzen betreffen nach Angaben der National Academies fast 1 von 5 Erwachsenen in den Vereinigten Staaten, doch die Cannabinoid-Pharmakologie hat keine universell verlässliche rezeptorbasierte Lösung hervorgebracht.
So betrachtet sind CB1 und CB2 besser als Kontrollknoten zu verstehen, die in Lipid-, Immun- und synaptische Schaltkreise eingebettet sind. Ihre Arzneimittel-Adressierbarkeit ist real. Ebenso ihre Fähigkeit, zu überraschen.
CB1 als Arzneiziel: Versprechen, Risiken und das Problem zentraler Nebenwirkungen
CB1 bleibt eines der verlockendsten Ziele in der Neuropharmakologie, weil es an einem strategischen Kontrollpunkt der synaptischen Signalübertragung sitzt. Die Rezeptorpharmakologie-Arbeiten von Allyn Howlett zeigten, dass cannabinoid-Wirkungen kein vages Membranphänomen waren, sondern rezeptorvermittelte Biologie, und die spätere Entdeckung von Anandamid durch Raphael Mechoulam und Lumír Hanuš gab diesem Rezeptor ein endogenes Signalsystem. Diese Geschichte ist bedeutsam. Ein Rezeptor, der die Freisetzung von Neurotransmittern in Schmerzschaltkreisen, Fütterungsschaltkreisen, Stresswegen und Belohnungsnetzwerken mitreguliert, erscheint als plausibler Ansatzpunkt für mehrere schwierige Erkrankungen zugleich. Sie schafft aber auch eine Falle: Wenn ein Rezeptor in so viele zentrale Hirnfunktionen eingebettet ist, bleibt eine breite Manipulation selten auf die gewünschte Wirkung beschränkt.
Dieses Problem ist nicht akademisch. Die World Health Organization schätzt, dass 200 Millionen Menschen im Jahr 2019 cannabis konsumierten, etwa 4 % der Weltbevölkerung im Alter von 15 bis 64 Jahren, sodass die CB1-Pharmakologie bereits auf Populationsebene durch pflanzliche cannabinoid-Wirkstoffe wirkt, insbesondere Delta-9-THC. Dennoch bleiben zugelassene cannabinoid-Arzneimittel selten: Die U.S. FDA gibt an, dass sie bis 2025 ein aus cannabis abgeleitetes Arzneimittelprodukt und drei cannabis-bezogene Arzneimittelprodukte zugelassen hat. Die Lücke zwischen weit verbreiteter Exposition und begrenzten zugelassenen Therapeutika sagt etwas Wichtiges über CB1 aus. Die Biologie ist überzeugend. Die Arzneientwicklung ist schwierig.
Therapeutische Begründung bei Schmerzen, Appetit und Neuropsychiatrie
Die Attraktivität beginnt mit der Frage, wo CB1 wirkt und was es tut. CB1 ist in vielen präsynaptischen Endigungen im zentralen Nervensystem stark exprimiert, wo endocannabinoid-Signale häufig bedarfsabhängig in postsynaptischen Zellen gebildet werden und rückwärts über die Synapse wandern, um die Transmitterfreisetzung zu hemmen. Dieses retrograde System kann je nach Zelltyp und Schaltkreis Glutamat-, GABA- und andere Signalströme dämpfen. Für Schmerzen ist das eine offensichtliche Begründung: Wenn fast 1 von 5 Erwachsenen in den Vereinigten Staaten mit chronischen Schmerzen lebt, wie die National Academies 2017 feststellten, wird ein Rezeptor, der nozizeptive Signalübertragung reduzieren und die Schmerzaffektivität verändern kann, zwangsläufig Aufmerksamkeit auf sich ziehen.
CB1 ist jedoch kein einfacher Analgesie-Schalter. In einigen Schaltkreisen kann die Hemmung der inhibitorischen GABA-Freisetzung nachgeschaltete Neurone enthemmen, statt sie zu beruhigen. In anderen überwiegt die glutamaterge Hemmung. Der Effekt hängt von der Synapse, dem Zustand des Netzwerks und dem Liganden ab. Endocannabinoid wie Anandamid und 2-AG sind kurzlebige, lokal gebildete Signale. Phytocannabinoide und synthetische Agonisten kommen mit anderer Kinetik, anderer Wirksamkeit und oft deutlich breiterer Rezeptorbindung über die Zeit an. Dieser Unterschied hilft zu erklären, warum „CB1-Aktivierung reduziert Schmerzen“ zu grob ist, um eine sichere klinische Entwicklung zu leiten.
Appetit ist eine zweite große Begründung. CB1-Signalgebung fördert die Nahrungsaufnahme und interagiert mit hypothalamischen und mesolimbischen Schaltkreisen, die Energiehomöostase mit Belohnung verbinden. Dies machte CB1-Antagonismus zu einer attraktiven Anti-Adipositas-Strategie und CB1-Agonismus oder indirekte Verstärkung zu einem potenziellen Ansatz bei Kachexie oder Appetitverlust. Die Logik war nicht irrational. Sie beruhte auf grundlegender Physiologie. Doch Appetit ist untrennbar mit Stimmung, Salienz und Stressverarbeitung verbunden, die ebenfalls durch CB1 geprägt werden.
Die Neuropsychiatrie ist der Bereich, in dem der Rezeptor am interessantesten und am gefährlichsten wird. CB1 moduliert Dopamin-relevante Schaltkreise, kortikale Hemmung, hippocampale Plastizität, Furchtlernen und Stressreaktivität. Das verleiht ihm theoretische Relevanz für Angststörungen, traumaassoziierte Erkrankungen, Depressionen, Substanzgebrauchsstörungen und psychotische Erkrankungen. The American Journal of Psychiatry argumentierte 2025, dass verzerrte CB1-Signalgebung eine plausible Strategie bei Schizophrenie sei, einer Störung, von der laut WHO weltweit etwa 24 Millionen Menschen betroffen sind. Die Bedeutung dieses Beitrags liegt nicht darin, dass ein CB1-Arzneimittel für Schizophrenie bereits bereitsteht. Sie liegt darin, dass sich das Feld von grober Agonisten- und Antagonisten-Pharmakologie hin zu pfadselektiver Pharmakologie bewegt und dabei auf den breiteren GPCR-Ansatz zurückgreift.
Warum die CB1-Adressierung wissenschaftlich attraktiv, klinisch aber schwierig ist
CB1 ist aus genau demselben Grund wissenschaftlich attraktiv, aus dem es klinisch schwierig ist: Es ist ein dichtes Signalzentrum. Auf der kanonischen Ebene koppelt CB1 vor allem an Gi/o-Proteine, reduziert die Adenylylcyclase-Aktivität, moduliert Ionenkanäle und hemmt die Neurotransmitterfreisetzung. Allein das würde bereits ausreichen, um es bedeutsam zu machen. Es rekrutiert aber auch beta-Arrestine, durchläuft Desensibilisierung und Internalisierung und kann durch allosterische Modulatoren sowie liganden-spezifische Konformationszustände geprägt werden. Die strukturelle Übersichtsarbeit in Frontiers in Chemical Biology aus dem Jahr 2026 macht dies für CB1 und CB2 klar: Ligandenselektivität und Wirksamkeit ergeben sich aus strukturellen Unterschieden auf Rezeptorebene, die Bindung, Signalausgabe und Rezeptorregulation beeinflussen. Ähnlich aussehende cannabinoid-Verbindungen können daher pharmakologisch sehr unterschiedliche Wirkungen entfalten.
Das ist nicht nur ein Detail der medizinischen Chemie. Es wirkt sich direkt auf Nebenwirkungen aus. Ein Ligand, der eine aktive Konformation stark antreibt, kann mehr Rezeptorinternalisierung, mehr Toleranz oder ein anderes Verhaltensprofil erzeugen als ein Ligand, der einen anderen Zustand stabilisiert. Eine PubMed-indexierte Studie aus 2025/2026 zur Subtypselektivität argumentierte zudem, dass endocannabinoid-Selektivität dynamisch und kein festes Schlüssel-Schloss-Ereignis sei, was die Vorstellung stützt, dass sich das Rezeptorverhalten mit dem Konformationskontext verändert. Arzneientwickler stehen also nicht vor einem einzelnen CB1-Ziel. Sie stehen vor einem beweglichen Ensemble.
Deshalb haben grobe CB1-Agonisten und -Antagonisten so häufig enttäuscht. Eine starke zentrale Agonisierung kann Gedächtnis, Aufmerksamkeit, psychomotorische Leistungsfähigkeit und Angstregulation beeinträchtigen und zugleich Belohnungswege so ansprechen, dass das Abhängigkeitsrisiko problematisch wird. Eine starke zentrale Antagonisierung kann zwar den Appetit senken, aber auch die gleichen stressdämpfenden und affektiven Schaltkreise stören, die endocannabinoide normalerweise stabilisieren. CB1 ist tief in Kognition, Belohnung und Stressregulation involviert. Jedes Arzneimittel, das dieses System zu stark in eine Richtung drängt, sollte bis zum Beweis des Gegenteils als riskant gelten.
Die Systembiologie stützt diese Vorsicht. Eine integrative Netzwerkanalyse aus 2025/2026 identifizierte CB1 und CB2 als hoch einflussreiche Knoten im endocannabinoid-System und verband die Rezeptorsignalgebung mit breiteren Stoffwechselwegen. Dieser Befund spricht gegen eine rezeptorzentrische Vereinfachung. Die Modulation von CB1 verschiebt nicht nur eine Synapse. Sie kann sich über endokrine, immunologische, metabolische und Verhaltensnetzwerke fortpflanzen.
Lehren aus zentral wirksamen versus peripher begrenzten Strategien
Die klarste Lehre aus der ersten Ära der CB1-Arzneientwicklung ist, dass zentrale Exposition häufig genau dort ist, wo Wirksamkeit und Risiko untrennbar werden. Zentral wirksame CB1-Agonisten können Schmerz- und Appetitschaltkreise ansprechen, aber dieselbe Hirngängigkeit erhöht die Wahrscheinlichkeit von intoxikationsähnlichen Effekten, Sedierung, kognitiver Beeinträchtigung und psychiatrischen Nebenwirkungen. Zentral wirksame CB1-Antagonisten zeigen den gegenteiligen Fehlermodus: Sie können metabolische Ziele erreichen, dabei aber inakzeptable Stimmungs- und Angsteffekte hervorrufen, weil sie auch den endocannabinoid-Tonus in limbischen Schaltkreisen blockieren.
Diese Geschichte ist der Grund, warum peripher begrenzte CB1-Liganden attraktiv wurden. Die Idee ist einfach: Wenn einige therapeutische Vorteile von CB1 in peripheren Geweben wie primären Afferenzen, Darm, Leber, Fettgewebe oder anderen Nicht-ZNS-Orten abhängen, dann könnte ein Arzneimittel, das aus dem Gehirn herausgehalten wird, nützliche Effekte erhalten und zentrale Nebenwirkungen verringern. Bei Schmerzen könnte dies bedeuten, periphere nozizeptive Mechanismen zu beeinflussen. Bei Stoffwechselerkrankungen könnte es bedeuten, Lipogenese zu reduzieren oder glukosebezogene Wege zu verändern, ohne Stimmung oder Kognition zu stören.
Die Logik ist schlüssig, aber sie ist keine magische Lösung. Erstens lassen sich periphere und zentrale CB1-Biologie in Krankheitszuständen nicht sauber trennen. Chronische Schmerzen, Nahrungsaufnahme und Stress umfassen alle Gehirn-Körper-Schleifen. Zweitens ist der Ausschluss über die Blut-Hirn-Schranke ein quantitatives, kein binäres Problem. Selbst geringe ZNS-Penetration kann relevant sein, insbesondere bei chronischer Gabe. Drittens kann ein wirklich peripherer CB1-Ligand klinisch dennoch scheitern, wenn die Zielerkrankung stark von zentralen Schaltkreisen abhängt oder kompensatorische Wege die Wirksamkeit abschwächen.
Die neuere Strategie lautet daher nicht bloß: „Bleibe außerhalb des Gehirns.“ Sie lautet: „Forme das Signal.“ Dazu gehören partielle Agonisten, neutrale Antagonisten statt inverser Agonisten, allosterische Modulatoren und verzerrte Liganden, die bestimmte nachgeschaltete Wege gegenüber anderen bevorzugen. Die Diskussion im American Journal of Psychiatry aus dem Jahr 2025 über verzerrte CB1-Signalgebung bei Schizophrenie passt hierher. Wenn ein Ligand ausgewählte therapeutische Signalwege erhalten und gleichzeitig beta-Arrestin-Rekrutierung, Rezeptordesensibilisierung oder unerwünschte Schaltkreiseffekte verringern kann, könnte CB1 handhabbarer werden. Könnte. Es gibt weiterhin keine Garantie, dass in Zellsystemen beobachtete Pfadverzerrung sich sauber auf menschliche therapeutische Fenster übertragen lässt.
Die heutige Sicht auf CB1 als Arzneiziel ist daher weder abwertend noch romantisch. CB1 ist attraktiv, weil es die synaptische Verstärkung genau in den Schaltkreisen reguliert, die Kliniker beeinflussen wollen. Es ist schwierig, weil diese Schaltkreise auch Gedächtnis, Affekt, Motivation und Stressresilienz steuern. Peripher begrenzte Liganden, selektive Signalstrategien und ein besseres strukturelles Verständnis haben die Designlogik verbessert. Sie haben das Grundproblem nicht beseitigt. Ein Rezeptor, der so zentral für die Hirnfunktion ist, toleriert selten eine grobe Pharmakologie.
CB2 als Arzneimittelziel: Neuroinflammation, Neurodegeneration und Immunmodulation
CB2 ist aus einem einfachen Grund zu einem der verlockendsten Ziele in der cannabinoid-Pharmakologie geworden: Es scheint Zugang zu Immun- und Glia-Biologie zu bieten, bei geringerem Risiko von Intoxikation, Gedächtnisbeeinträchtigung und Missbrauchspotenzial, die klassisch mit einer starken CB1-Aktivierung verbunden sind. Diese Attraktivität ist weit mehr als nur für das akademische Rezeptor-Mapping von Bedeutung. Die Weltgesundheitsorganisation schätzte, dass im Jahr 2019 200 Millionen Menschen cannabis konsumierten, also 4 % der Weltbevölkerung im Alter von 15 bis 64 Jahren, und die U.S. FDA führt auch 2025 weiterhin nur ein aus cannabis gewonnenes Arzneimittel und drei cannabisbezogene Arzneimittelprodukte als zugelassen. Das Interesse ist groß. Die Übertragung in die Praxis ist schwierig.
Die ältere Kurzformel lautete: CB1 ist der Gehirnrezeptor und CB2 ist der Immunrezeptor. Das ist heute zu grob, um nützlich zu sein. Die 2026 in Frontiers in Behavioral Neuroscience veröffentlichte Übersichtsarbeit, ausdrücklich als „ein Update der letzten 3 Jahre“ konzipiert, behandelt CB2 als relevant für Erkrankungen des zentralen Nervensystems, gerade weil es an neuroinflammatorischen und neurodegenerativen Mechanismen beteiligt ist, einschließlich Mikroglia-Aktivierung, Zytokin-Signalisierung und der Kommunikation zwischen Neuronen und Glia. Der entscheidende Wandel besteht nicht darin, dass CB2 plötzlich überall im Gehirn in großer Menge vorhanden wäre. Das ist nicht der Fall. Der Wandel besteht darin, dass eine niedrige basale Expression in vielen ZNS-Regionen nicht bedeutet, dass keine funktionelle Relevanz besteht, insbesondere wenn Krankheitszustände die Rezeptorexpression, den Transport und die Signalübertragung verändern können.
Warum CB2 oft als sichereres Ziel angesehen wird
Das Sicherheitsargument für CB2 beginnt bei der Verteilung, endet dort aber nicht. CB2 ist in Immunzellen angereichert und wird häufig in aktivierter Mikroglia, infiltrierenden Makrophagen und anderen entzündlichen Zellpopulationen hochreguliert. Im Gegensatz dazu ist CB1 dicht an präsynaptischen Endigungen im Kortex, Hippocampus, in den Basalganglien und im Kleinhirn exprimiert, wo es die Freisetzung von Neurotransmittern stark beeinflusst. Dieser anatomische Unterschied ist wichtig, weil ein Ligand, der hauptsächlich CB2 anspricht, weniger wahrscheinlich das klassische psychoaktive Profil nachbildet, das mit CB1 verbunden ist.
Weniger wahrscheinlich ist nicht dasselbe wie unmöglich. Selektivität ist ein bewegliches Ziel. Die 2026 in Frontiers in Chemical Biology veröffentlichte Übersichtsarbeit betont, dass die Selektivität für „CB1 und CB2“ von strukturellen Unterschieden abhängt, die Bindung, Wirksamkeit und Rezeptorregulation verändern, und eine kürzlich in PubMed indexierte Studie zur Subtyp-Selektivität argumentiert, dass die Unterscheidung von endocannabinoid zwischen Rezeptorsubtypen eher konformationelle Dynamiken widerspiegelt als ein festes Schloss-und-Schlüssel-Modell. In der Praxis bedeutet das, dass eine Verbindung in einem Assay CB2-selektiv erscheinen und sich in einem anderen deutlich weniger sauber verhalten kann, insbesondere wenn Rezeptordichte, Membranumgebung und nachgeschaltete Transduktoren variieren. Derselbe Ligand kann in einem Zelltyp eine G-Protein-Signalisierung bevorzugen, in einem anderen Beta-Arrestine rekrutieren und unter entzündlichen Bedingungen unterschiedliche Desensibilisierungs- oder Internalisierungskinetiken zeigen.
Diese mechanistische Komplexität stärkt paradoxerweise das Argument, CB2 sorgfältig zu verfolgen. Wenn das therapeutische Ziel darin besteht, die Zytokinfreisetzung der Mikroglia zu reduzieren, die Rekrutierung peripherer Immunzellen zu begrenzen oder glialen metabolischen Stress zu verändern, ohne die kortikale synaptische Übertragung stark zu beeinträchtigen, dann bleibt CB2 der plausiblere erste Rezeptor. CB1-biased signaling könnte, wie in dem 2025 in American Journal of Psychiatry erschienenen Artikel zu Schizophrenie ausgeführt, den therapeutischen Fensterbereich von auf CB1 ausgerichteten Arzneimitteln eines Tages erweitern. Das ist jedoch ein Argument für intelligentere CB1-Pharmakologie, nicht ein Grund, die geringere psychoaktive Belastung CB2-fokussierter Ansätze zu ignorieren.
Sicherer sollte jedoch nicht mit wirksam verwechselt werden. Ein Arzneimittel kann eine offensichtliche Intoxikation vermeiden und dennoch versagen, weil der Rezeptor im Krankheitsprozess zu spärlich, zu variabel oder zu kurzzeitig exprimiert ist, um einen relevanten klinischen Effekt zu erzeugen. Das ist in der Entwicklung von Arzneimitteln gegen Neuroinflammation wiederholt geschehen, und zwar nicht nur in der cannabinoid-Wissenschaft.
Evidenz und Grenzen in ZNS-Krankheitsmodellen
Die Übersichtsarbeit in Frontiers in Behavioral Neuroscience zeigt, warum CB2 in Modellen von Alzheimer-Krankheit, Parkinson-Krankheit, Multipler Sklerose, Schädel-Hirn-Trauma, Schlaganfall und neuropathischem Schmerz immer wieder auftaucht: Diese Erkrankungen umfassen alle entzündliche Signalisierung, gliale Reaktivität, oxidativen Stress und fortschreitende Gewebeschädigung. In vielen Nagetierstudien reduzieren CB2-Agonisten proinflammatorische Mediatoren wie TNF-α, IL-1β und IL-6, verschieben Mikroglia in weniger schädliche Phänotypen oder dämpfen die Leukozyteninfiltration. Manchmal geht dies mit verbessertem motorischem Verhalten, reduzierter Läsionsgröße oder dem Erhalt synaptischer Marker einher. Das sind reale Signale, keine Artefakte, die man abtun sollte.
Die Datenlage ist jedoch uneinheitlich. Die Effekte hängen oft vom Zeitpunkt ab. Ein CB2-Agonist, der während der frühen Entzündungswelle nach einer Verletzung verabreicht wird, kann etwas völlig anderes bewirken als derselbe Agonist in einer Phase chronischer Degeneration. Auch die Dosis spielt eine Rolle, ebenso die Ligandenklasse. Endocannabinoide wie 2-AG und Anandamid sind nicht mit Phytocannabinoiden oder synthetischen CB2-bevorzugten Agonisten austauschbar. Ihre lokalen Konzentrationen, ihr metabolischer Abbau und ihre Off-Target-Wirkungen unterscheiden sich. Gleiches gilt für ihre Präferenz für bestimmte Signalwege.
Die Krankheitsmodelle selbst sind eine weitere Grenze. Eine transgene Maus mit Alzheimer-Pathologie und reaktiver Mikroglia ist nicht die Alzheimer-Krankheit bei einem 74-jährigen Menschen mit gemischtem vaskulärem, inflammatorischem und proteostatischem Versagen. Experimentelle Autoimmunenzephalomyelitis ist für die Biologie der Multiplen Sklerose nützlich, bildet aber nur einen Teil der menschlichen Erkrankung ab. Positive präklinische Ergebnisse begründen daher Plausibilität, nicht Beweis.
Hinzu kommt ein anhaltendes Messproblem in der CB2-Biologie. Die Spezifität von Antikörpern war lange ein Schwachpunkt, wodurch Aussagen zur Rezeptorlokalisation weniger vertrauenswürdig sind, als viele Arbeiten nahelegen. Niedrige Expressionsniveaus im gesunden Gehirn, induzierbare Expression bei Krankheit und Speziesunterschiede erschweren die Interpretation zusätzlich. Manche berichteten Befunde zu „neuronalem CB2“ mögen in spezifischen Kontexten real sein, doch breite Aussagen über eine weit verbreitete konstitutive neuronale CB2-Expression sind weiterhin weniger gesichert, als die Fachwelt einst annahm.
Hier ist die Signalisierung ebenso wichtig wie die Anatomie. CB2 ist ein Gi/o-gekoppelter GPCR, doch diese Kurzform verdeckt wichtige Variabilität: Hemmung der Adenylylcyclase, Modulation von MAPK-Signalwegen, Effekte auf Ionenkanäle, Rekrutierung von Beta-Arrestin, Rezeptordesensibilisierung und Internalisierung können alle das Netto-Biologieergebnis verändern. Ein Ligand, der in kultivierter Mikroglia die Zytokinfreisetzung reduziert, kann in vivo versagen, wenn er Rezeptoren rasch desensibilisiert oder ein ungünstiges, arrestin-dominiertes Programm auslöst. Ein geringeres Intoxikationsrisiko löst diese pharmakologischen Probleme nicht.
Was die neuere Literatur für zukünftige Therapien nahelegt
Die neuere Literatur weist weg von der alten Suche nach einem einzelnen „CB2-Agonisten gegen Hirnentzündung“ hin zu einer selektiveren, kontextabhängigen Strategie. Sowohl die strukturelle Übersichtsarbeit von 2026 als auch die Studie zur Subtyp-Selektivität von 2025/2026 stützen dieselbe Lehre: Die Rezeptoransprache wird von Konformationszustand, Wirksamkeitsprofil und Gewebekontext abhängen, nicht nur von der nominalen Affinität zum Subtyp. Die Arzneimittelentwicklung bewegt sich in Richtung Liganden, die funktionell selektiv sind, nicht nur in der Bindung.
Das dürfte drei Dinge bedeuten. Erstens werden zukünftige CB2-Arzneimittel bessere Signaturprofile der Signalübertragung benötigen, einschließlich G-Protein- versus Beta-Arrestin-Bias, Rezeptorverweilzeit und zelltypspezifischer Effekte in menschlichen Systemen und nicht nur in immortalisierten Assay-Linien. Zweitens wird eine Krankheitsstratifizierung wichtig sein. Eine auf CB2 ausgerichtete Therapie könnte dort am besten wirken, wo entzündliche Verstärkung ein wesentlicher Treiber und nicht nur ein Begleitphänomen der Pathologie ist. Drittens ist ein kombinatorisches Denken unvermeidlich. Eine in einer kürzlich in PubMed indexierten Studie veröffentlichte integrative Netzwerkanalyse identifizierte CB1 und CB2 als einflussreiche Knoten in breiteren endocannabinoid- und Stoffwechselwegen, was der gelebten Realität von ZNS-Erkrankungen entspricht: Entzündung, mitochondrialer Stress, Lipidsignalisierung und synaptische Dysfunktion sind eng verflochten.
Unter spezifischen entzündlichen Bedingungen kann die Modulation von CB2 schädliche Immunsignale verringern, ohne einen Großteil der mit CB1 verbundenen psychoaktiven Last zu verursachen.Preliminary evidence
Das macht CB2 nicht zu einer Sackgasse. Es macht es zu einem Ziel, das wahrscheinlich eher in die Präzisionspharmakologie als in die Rezeptormythologie gehört. Das derzeit stärkste Argument lautet nicht „CB2-Aktivierung behandelt Neurodegeneration“. Es ist enger gefasst: Unter spezifischen entzündlichen Bedingungen, in spezifischen Zellpopulationen und mit den richtigen Ligandeneigenschaften kann die Modulation von CB2 schädliche Immun-Signale reduzieren und dabei einen Großteil der mit CB1 verbundenen psychoaktiven Belastung vermeiden. Das ist eine plausible therapeutische Hypothese. Es ist noch keine klinisch validierte Regel.
Cannabis-Verbindungen an CB1 und CB2: THC, CBD, Minor-Cannabinoide und synthetische Liganden
cannabis wirkt nicht über eine einzelne chemische Substanz oder einen einzelnen Rezeptormechanismus. Dieser Punkt klingt offensichtlich, doch die öffentliche Diskussion reduziert die Pharmakologie weiterhin auf ein falsches Paar: THC entspricht CB1, CBD entspricht CB2. Die Literatur stützt diese Vereinfachung nicht. cannabis enthält viele Phytocannabinoide sowie Terpene, Flavonoide und andere Bestandteile, während der Körper eigene Endocannabinoide wie Anandamid und 2-Arachidonylglycerol bildet, die durch Arbeiten im Umfeld von Raphael Mechoulam, Lumír Hanuš und anderen identifiziert wurden. Darüber hinaus stammt der Großteil dessen, was Wissenschaftler über Rezeptoraktivierung, Wirksamkeit, Selektivität, Desensibilisierung und Signal-Bias wissen, aus synthetischen Liganden, die als Sonden oder Arzneikandidaten entwickelt wurden, und nicht allein aus Pflanzenbestandteilen.
Diese Unterscheidung ist wichtig, weil CB1 und CB2 keine passiven Schlösser sind, die nur auf einen Cannabinoid-Schlüssel warten. Sie sind Klasse-A-GPCRs mit mehreren Konformationszuständen, Kopplungspräferenzen und regulatorischen Verläufen. Eine strukturelle Übersichtsarbeit von 2026 in Frontiers in Chemical Biology macht dies deutlich: Die Ligandenselektivität an CB1 und CB2 hängt von strukturellen Unterschieden auf Rezeptorebene ab, die Bindung, Wirksamkeit und Rezeptorregulation verändern, und nicht nur davon, ob ein Molekül einen der beiden Rezeptoren „trifft“. Eine kürzlich in PubMed indexierte Studie zur Subtypselektivität argumentiert ebenfalls, dass die Präferenz von Endocannabinoiden für CB1 gegenüber CB2 aus konformationeller Dynamik und nicht aus einer einfachen Schlüssel-Schloss-Passung resultiert. Wenn zwei Cannabinoide chemisch ähnlich aussehen, aber unterschiedliche Effekte im Gehirn, im Immungewebe oder in Schmerzschaltkreisen hervorrufen, ist das kein Randaspekt. Es ist die zentrale Pharmakologie.
Angesichts der Schätzung der WHO, dass im Jahr 2019 200 Millionen Menschen cannabis konsumierten, also etwa 4 % der Weltbevölkerung im Alter von 15 bis 64 Jahren, sind diese Unterschiede keine akademische Nebensache. Sie prägen Intoxikation, therapeutische Aussagen, Sicherheit und die Frage, warum manche rezeptorzielgerichteten Arzneimittel Erfolg haben, während andere scheitern.
Wie THC CB1 aktiviert und zu psychoaktiven Effekten beiträgt
Delta-9-Tetrahydrocannabinol, oder THC, ist nach wie vor die zentrale Substanz zum Verständnis der cannabis-Intoxikation, weil es CB1 direkt im Zentralnervensystem aktiviert. Das ist die klarste Erklärung auf Rezeptorebene für die charakteristischen psychoaktiven Effekte von cannabis, auch wenn „psychoaktiv“ mehrere voneinander trennbare Wirkungen umfasst: veränderte Zeitwahrnehmung, Belohnung, Gedächtnisstörung, Anxiogenese bei einigen Nutzern und beeinträchtigte motorische Koordination. Allyn Howletts Arbeiten zur Rezeptorpharmakologie trugen dazu bei, dieses CB1-zentrierte Modell zu etablieren, und es ist bis heute grundlegend.
Doch „THC aktiviert CB1“ ist nur der Anfang. THC ist kein perfekter Einschaltknopf. Es wird allgemein als partieller Agonist an CB1 beschrieben, was bedeutet, dass seine Wirksamkeit von Rezeptordichte, Gewebekontext und dem verwendeten Signalassay abhängt. In CB1-reichen Regionen wie Kortex, Hippocampus, Basalganglien und Kleinhirn kann das ausreichen, um starke zentrale Effekte hervorzurufen. Auf synaptischer Ebene ist CB1 oft präsynaptisch lokalisiert; dort hemmt seine Aktivierung die Neurotransmitterfreisetzung. Das bedeutet, dass THC in einem Schaltkreis die Glutamatfreisetzung dämpfen, in einem anderen die GABA-Freisetzung reduzieren und je nach exprimierenden Zellen und Zeitpunkt gegensätzliche Netzwerk-Effekte hervorrufen kann.
Deshalb lässt sich die Rezeptorlokalisation nicht auf „CB1 im Gehirn“ reduzieren. CB1 ist zwar im Gehirn reichlich vorhanden, aber nicht gleichmäßig verteilt, nicht statisch und nicht in allen Neuronentypen funktionell identisch. Das Ergebnis ist, dass die Effekte von THC schaltkreisabhängig sind. Euphorie und Verstärkung gehören ebenso dazu wie kurzfristige Gedächtnisstörung und bei vulnerablen Personen psychose-relevante Signalwege.
Dieser letzte Punkt ist wichtiger geworden, nicht weniger. Ein Artikel von 2025 im American Journal of Psychiatry argumentierte, dass CB1-Bias-Signaling eine plausible therapeutische Strategie bei Schizophrenie sei, gerade weil CB1-Biologie nicht als eindimensionale Rezeptoraktivierung verstanden werden kann. Schizophrenie betrifft laut WHO weltweit etwa 24 Millionen Menschen, und jede ernsthafte Diskussion von THC an CB1 muss berücksichtigen, dass dieselbe Rezeptorfamilie, die an Intoxikation beteiligt ist, auch über biased agonism, beta-arrestin-Rekrutierung und pfadselektives Wirkstoffdesign untersucht wird. THC kann Gi/o-gekoppelte Signale auslösen, doch CB1 unterliegt auch Phosphorylierung, Desensibilisierung und Internalisierung, mit Folgen für Toleranz und nachgeschaltete Antworten. Dosis und Zeitpunkt sind entscheidend. Wiederholte Exposition verändert das System.
Synthetische CB1-Liganden haben dies schon vor langer Zeit deutlich gemacht. Verbindungen wie CP55,940 und WIN55,212-2 sind potente Forschungsagonisten, die in Laborassays oft stärkere oder klarere Rezeptorantworten hervorrufen als THC. Rimonabant, ein CB1-Inversagonist, zeigte die entgegengesetzte Richtung: Das Blockieren von CB1 oder das Absenken seiner Aktivität unter das basale Niveau konnte Appetit und Körpergewicht reduzieren, doch psychiatrische Nebenwirkungen brachten das Arzneimittel zu Fall. Diese klinische Geschichte ist eine Warnung. Selektive CB1-Targetierung kann pharmakologisch elegant und dennoch medizinisch enttäuschend sein.
Warum CBD nicht in ein einfaches CB1/CB2-Agonistenmodell passt
CBD ist das Cannabinoid, das am häufigsten in Mythen geglättet wird. Es wird routinemäßig als „das nicht-intoxizierende, das über CB2 wirkt“ oder als direkter Gegenpol zu THC dargestellt. Keine dieser Aussagen ist korrekt.
| Verbindungsklasse oder Beispiel | Wie der Artikel das Rezeptorverhalten beschreibt | Zentrale Schlussfolgerung |
|---|---|---|
| THC | Partieller Agonist an CB1 und CB2; zentral für die Intoxikation über CB1 | CB1-Bindung hilft, psychoaktive Wirkungen zu erklären, das Ergebnis bleibt jedoch kontextabhängig |
| CBD | Kein einfacher CB1- oder CB2-Agonist; kann allosterisch oder indirekt wirken | CBD ist kein einfaches Spiegelbild von THC an CB2 |
| Minor-Cannabinoide | Berichtete Wirkungen variieren je nach Testsystem und Konzentration | Das Rezeptorverhalten eines einzelnen Cannabinoids nicht auf alle verallgemeinern |
| Synthetische Liganden | Oft verwendet, um Wirksamkeit, Selektivität und Bias zu bestimmen | Sondenverbindungen können sich stark von Pflanzen-Cannabinoiden unterscheiden |
CBD verhält sich nicht wie ein einfacher CB1- oder CB2-Agonist, wie THC an CB1. Seine Affinität an den orthosterischen Bindungsstellen von CB1 und CB2 ist relativ gering, und ein Großteil seiner Pharmakologie scheint indirekte, allosterische oder nicht-cannabinoide Rezeptorwirkungen zu umfassen. Je nach untersuchtem System wurde CBD als negativer allosterischer Modulator an CB1 beschrieben, was verändern könnte, wie THC oder Endocannabinoide Signale auslösen, ohne den Rezeptor schlicht an- oder auszuschalten. Diese Unterscheidung ist entscheidend. Die Modulation von Rezeptorform und Signal-Effizienz ist nicht dasselbe wie klassische Agonistik.
CBD interagiert außerdem mit Zielstrukturen jenseits von CB1 und CB2, darunter TRPV1, 5-HT1A, PPAR-gamma, Adenosin-bezogene Signalwege und Enzyme, die an der endocannabinoiden Tonusregulation beteiligt sind. Einige Effekte können eher auf veränderte Anandamid-Signale als auf direkte Rezeptorstimulation zurückgehen. Das hilft zu erklären, warum das klinische Profil von CBD nicht sauber mit der alten Rezeptor-Kurzformel übereinstimmt. Die U.S. FDA weist darauf hin, dass sie bis 2025 ein cannabis-abgeleitetes Arzneimittelprodukt und drei cannabis-bezogene Arzneimittelprodukte zugelassen hat; die am besten belegte medizinische Anwendung von CBD liegt bei Epilepsie, einer Erkrankung, von der weltweit etwa 50 Millionen Menschen betroffen sind, und dieser therapeutische Weg lässt sich nicht durch ein Karikaturmodell von „CBD bindet CB2 und reduziert Entzündung“ erklären.
Selbst für CB2 ist das Bild breiter als die ältere periphere Darstellung. Eine Übersichtsarbeit von 2026 in Frontiers in Behavioral Neuroscience beschrieb „ein Update der letzten 3 Jahre“, in dem CB2-Signalgebung durch Verbindungen zu neuroinflammatorischen und neurodegenerativen Mechanismen mehr Aufmerksamkeit im Kontext von Erkrankungen des zentralen Nervensystems erhielt. Das bedeutet nicht, dass CBD einfach ein CB2-Arzneimittel ist. Es bedeutet, dass der Rezeptor selbst eine größere funktionelle Reichweite hat, als die öffentliche Darstellung vermuten lässt.
Die Bedeutung der Trennung zwischen Pflanzenverbindungen und Rezeptormythen
Phytocannabinoide sind nicht austauschbar, und sie sind auch nicht mit synthetischen Liganden austauschbar. Minor-Cannabinoide wie Cannabigerol (CBG), Cannabinol (CBN), Tetrahydrocannabivarin (THCV) und Cannabichromen (CBC) wurden als Interaktionspartner von CB1 und CB2 beschrieben, wobei die Effekte je nach Assay und Konzentration variieren. Einige zeigen schwache Agonistik, einige partielle Agonistik, einige Antagonismus unter bestimmten Bedingungen und einige bedeutsame Aktivität an Nicht-Cannabinoid-Zielstrukturen. THCV ist ein gutes Beispiel: In einigen Systemen wurde es bei niedrigen Dosen als CB1-Antagonist oder neutraler Antagonist beschrieben, bei höheren Dosen dagegen als partieller Agonist. Das ist keine Inkonsistenz um ihrer selbst willen. Es spiegelt kontextabhängige Rezeptorpharmakologie wider.
Forschungswerkzeuge schärfen diesen Punkt. Endocannabinoide, Phytocannabinoide und synthetische Liganden können zwar alle „CB1 und CB2“ aktivieren, stabilisieren aber nicht dieselben Rezeptorkonformationen und erzeugen nicht dieselbe Bias-Verteilung über G-Proteine und beta-Arrestine hinweg. Die strukturelle Übersichtsarbeit von 2026 und aktuelle in PubMed indexierte Netzwerkanalysen drängen das Feld gleichermaßen weg von Rezeptormythologie und hin zu Systempharmakologie. Rezeptoren liegen in zelltypspezifischen Signalnetzwerken, Stoffwechselwegen und Trafficking-Maschinerien eingebettet. Das klinische Profil eines Liganden ergibt sich aus diesem Gesamtzusammenhang, nicht aus einem Etikett wie „CB2-entzündungshemmend“ oder „CBD gleicht THC aus“.
Für ein cannabis-Wiki ist genau das die wichtigste Linie: THC hat primäre Relevanz für die CB1-vermittelte Intoxikation, doch die cannabis-Pharmakologie endet dort nicht, und CBD ist kein einfaches CB2-Spiegelbild von THC. Pflanzenverbindungen beginnen die Geschichte. Rezeptorzustand, Gewebeverteilung, Wirksamkeit, Bias und Regulation entscheiden, wie sie endet.
Schizophrenie, Psychoserisiko und was die CB1-Biologie bedeutet und nicht bedeutet
Warum Schizophrenie in die Rezeptor-Debatte einfließt
Schizophrenie ist hier von Bedeutung, weil CB1 nicht einfach ein weiterer Rezeptor auf einer langen Zielliste ist. Er ist einer der häufigsten G-Protein-gekoppelten Rezeptoren im Gehirn und an präsynaptischen Endigungen lokalisiert, wo er die Transmitterfreisetzung dämpfen und die Netzwerkaktivität in Schaltkreisen umgestalten kann, die bereits mit Psychosen in Verbindung gebracht werden: Kortex, Hippocampus, Striatum, Amygdala. Wenn ein Rezeptor so nahe an der Kontrolle glutamaterger, GABAerger, dopaminbezogener Schaltkreise und der Stressreaktivität sitzt, werden psychiatrische Forschende aufmerksam.
Das Ausmaß der öffentlichen Gesundheit macht daraus mehr als eine akademische Frage. Die Weltgesundheitsorganisation schätzt, dass cannabis im Jahr 2019 von 200 Millionen Menschen verwendet wurde, also von 4 % der Weltbevölkerung im Alter von 15 bis 64 Jahren. Die WHO schätzt außerdem, dass Schizophrenie etwa 24 Millionen Menschen weltweit betrifft, also ungefähr 1 von 300 Menschen. Das sind keine kleinen, isolierten Populationen. Sie überschneiden sich in realen Kliniken, in Notaufnahmen und in longitudinalen Kohortenstudien. Jede Rezeptorbiologie, die Psychoserisiko, die Entwicklung antipsychotischer Arzneimittel oder beides erhellen könnte, verdient ernsthafte Prüfung.
Das ist der translationale Hintergrund für den 2025 in American Journal of Psychiatry veröffentlichten Artikel, der argumentiert, dass ein biased signaling an CB1 eine plausible therapeutische Strategie für Schizophrenie ist. Die Kernaussage ist einfach zu formulieren und leicht zu missbrauchen. Einfach zu formulieren: CB1 verhält sich nicht wie ein einfacher An-Aus-Schalter, und unterschiedliche Liganden können unterschiedliche Rezeptorkonformationen stabilisieren und dadurch das Signal in Richtung spezifischer intrazellulärer Signalwege verschieben oder von ihnen weglenken. Leicht zu missbrauchen: Wenn CB1 für die Forschung zur Schizophreniebehandlung relevant ist, springen manche Leser zu der falschen Behauptung, „cannabinoid Aktivierung hilft bei Schizophrenie“ oder cannabis sei damit per se medizinisch. Der Artikel stützt diesen Schluss nicht.
Historisch blieb die cannabinoid-Wissenschaft in vereinfachenden Kurzformen stecken. CB1 wurde als der „Gehirnrezeptor“ dargestellt, CB2 als der „Immunrezeptor“, und psychoaktive Effekte wurden nahezu ausschließlich auf die CB1-Besetzung zurückgeführt. Diese Einordnung war immer unvollständig, und im Jahr 2025 ist sie offensichtlich zu grob. Die grundlegenden Arbeiten von Allyn Howlett etablierten die Pharmakologie der cannabinoid-Rezeptoren, während Mechoulam und Hanus mit Anandamid die endocannabinoid-Biologie mitprägten. Seither hat sich die Rezeptorpharmakologie von der Rezeptorpräsenz zum Rezeptorzustand entwickelt. Dieser Wandel ist für Schizophrenie bedeutsam, weil Pathophysiologie nicht allein durch Rezeptorexpression verursacht wird. Sie entsteht aus Timing, Zelltyp, Schaltkreis-Kontext und intrazellulärer Antwort.
Die neuere Literatur stärkt diesen Punkt. Eine 2026 in Frontiers in Chemical Biology veröffentlichte Übersicht erklärt, dass strukturelle Unterschiede zwischen CB1 und CB2 die Ligandenbindung, Wirksamkeit, Signalübertragung und Rezeptorregulation prägen. Eine 2025/2026 in PubMed indexierte Studie zur Subtyp-Selektivität argumentiert, dass die Selektivität von endocannabinoid durch Konformationsdynamik und nicht durch ein statisches Schlüssel-Schloss-Modell entstehen kann. Eine weitere 2025/2026 veröffentlichte Netzwerk-Analyse identifiziert CB1 und CB2 als hochgradig einflussreiche Knoten im gesamten endocannabinoid-System, die mit Stoffwechsel- und Signalwegen verknüpft sind und nicht mit isolierten Rezeptor-Boxen. Für die Schizophrenieforschung bedeutet das, dass man nicht nur fragen sollte: „Bindet eine Substanz an CB1?“, sondern auch: „Welche CB1-Zustände in welchen Zellen unter welchen Expositionsbedingungen?“
Daraus erwächst die klinische Ernsthaftigkeit. Nicht aus Schlagworten über Rezeptoren. Aus mechanistischer Präzision.
Biased signaling versus grobe Rezeptoraktivierung
Eine grobe Aktivierung von CB1 ist ein schlechtes Modell für therapeutisches Denken. Sie reduziert zu viele Variablen auf ein Wort: Aktivierung. Endogene Liganden wie Anandamid und 2-AG, phytocannabinoids wie Delta-9-THC und synthetische Agonisten verhalten sich an CB1 nicht identisch, selbst wenn sie alle als Liganden gelten. Sie können sich in Affinität, Wirksamkeit, Verweildauer, regionaler Exposition und Signalweg-Bias unterscheiden. Sie können sich auch darin unterscheiden, wie stark sie Gi/o-Proteine im Vergleich zu Beta-Arrestinen rekrutieren, wie schnell sie Desensibilisierung auslösen und wie viel Rezeptor-Internalisierung nach wiederholter Gabe folgt.
Diese Unterscheidung ist zentral für das Schizophrenie-Argument in AJP. Ein Ligand, der CB1 in Richtung eines intrazellulären Programms verschiebt, muss einem Liganden nicht ähneln, der eine breite Rezeptoraktivierung in vielen Hirnregionen auslöst. In der GPCR-Pharmakologie kann dieser Unterschied ein nützliches Arzneimittel von einem unzumutbaren trennen. CB1 ist für dieses Problem besonders empfindlich, weil derselbe Rezeptor in einer Synapse exzitatorische Transmission senken, in einer anderen inhibitorische Transmission unterdrücken und nachgeschaltete Plastizität in funktionell nicht äquivalenter Weise verändern kann. Kleine molekulare Unterschiede können große Konsequenzen auf Systemebene haben.
THC ist das offensichtliche Beispiel dafür, warum „CB1-Ziel“ nicht gleich „CB1-Therapie“ ist. THC ist ein phytocannabinoid mit partiellen Agonisten-Eigenschaften an CB1, aber die erlebten und klinischen Effekte von THC sind keine reine Abbildung eines einzelnen Signalwegs. Die Dosis ist wichtig. Das Alter bei Exposition ist wichtig. Wiederholte Exposition ist wichtig. Ebenso wichtig sind das Verhältnis von THC zu anderen Bestandteilen und die Frage, ob eine Person eine psychiatrische Vulnerabilität hat. Akute intoxikationsähnliche Effekte, Angst, Wahrnehmungsveränderungen und vorübergehende psychotische Symptome bei manchen Konsumierenden bedeuten nicht, dass alle auf CB1 zielenden Therapeutika zum Scheitern verurteilt sind. Sie zeigen jedoch, dass eine ungezielte Stimulation von CB1 kein Abkürzungsweg zu einer antipsychotischen Behandlung ist.
Biased agonism ist gerade deshalb attraktiv, weil es diese Grobheit vermeiden will. Die therapeutische Hoffnung lautet nicht „mehr CB1“. Sie lautet „das richtige CB1-Signalprofil“. Das kann bedeuten, Signalwege zu bevorzugen, die mit Schaltkreis-Stabilisierung verbunden sind, während Wege begrenzt werden, die mit ungünstigen kognitiven oder psychotomisierenden Effekten, Toleranz oder Rezeptor-Downregulation verknüpft sind. Ob dies bei Patienten erreichbar ist, bleibt offen. Das Konzept ist plausibel. Es ist kein klinisch bewiesener Erfolg.
CB2 gehört ebenfalls in dieses Bild, allerdings nicht als einfache Lösung. Die 2026 in Frontiers in Behavioral Neuroscience veröffentlichte Übersicht beschreibt „ein Update der letzten 3 Jahre“, in dem CB2-Signaling wegen neuroinflammatorischer und neurodegenerativer Zusammenhänge verstärkt Beachtung fand. Das schwächt die alte Behauptung, CB2 sei für Hirnerkrankungen irrelevant. Aber es rehabilitiert die alte Binarität nicht, indem es sie einfach umkehrt. Schizophrenie ist nicht nur Entzündung, und CB2 ist nicht nur ein antiinflammatorischer Schalter. Jedes ernsthafte translationale Modell muss Interaktionen zwischen Neuronen und Glia, Immun-Signaling und Schaltkreisdysfunktion gemeinsam berücksichtigen.
Was nicht gefolgert werden sollte
- Kein Wirksamkeitsbeweis Dass CB1 ein Arzneimittelziel ist, bedeutet nicht, dass Cannabiskonsum vor Schizophrenie oder Psychose schützt.
- Keine Widerlegung des Risikos Mechanistische Rezeptorstudien beseitigen nicht die epidemiologische Sorge über hohe THC-Exposition bei vulnerablen Personen.
- Keine Austauschbarkeit Endocannabinoide, Phytocannabinoide und synthetische Liganden sollten nicht als dieselbe Art von Exposition behandelt werden.
- Keine pauschale Zustimmung Bestehende FDA-Zulassungen beantworten Fragen zur psychiatrischen Sicherheit oder Wirksamkeit für einen breiten Cannabinoidgebrauch nicht abschließend.
Was Leser aus mechanistischen Studien nicht ableiten sollten
Erstens sollten Leser nicht daraus schließen, dass cannabis-Konsum schützend gegen Schizophrenie oder Psychose wirke, nur weil CB1 ein therapeutisches Ziel ist. Logik von Wirkstoff und Zielstruktur funktioniert nicht so. Beta-Rezeptoren sind Arzneimittelziele; das bedeutet nicht, dass jedes Stimulans eine Therapie ist. Opioid-Rezeptoren sind Arzneimittelziele; das bedeutet nicht, dass jede Opioid-Agonisierung nützlich ist. Dasselbe Prinzip gilt hier.
Zweitens heben mechanistische Studien epidemiologische Bedenken nicht auf. Forschung zu Rezeptor-Bias und Liganden-Design steht neben, nicht gegen, der Evidenz, dass cannabis-Exposition das Psychoserisiko bei manchen Menschen verschlechtern kann, insbesondere bei Produkten mit hohem THC-Gehalt, früher Initiierung oder starkem Konsum. Ein Rezeptormechanismus kann erklären, warum einige Substanzen für bessere Ergebnisse entwickelt werden könnten. Er rechtfertigt keine pauschalen öffentlichen Aussagen, wonach „CB1-Aktivierung gut für Schizophrenie ist“.
Drittens sollten Leser endocannabinoid, phytocannabinoids und synthetische Liganden nicht als austauschbar betrachten. Anandamid ist nicht THC. Ein peripher beschränkter CB1-Modulator ist nicht gerauchtes cannabis. Eine niedereffiziente, signalweg-biased experimentelle Substanz ist kein hoch exponiertes Konsumprodukt. Diese Kategorien sind wichtig, weil das Rezeptorverhalten von Ligandenklasse, Dosis, Applikationsweg, Timing und Gewebeexposition abhängt.
Schließlich entscheidet das Vorhandensein zugelassener cannabis-bezogener Arzneimittel die psychiatrischen Fragen nicht. Nach Angaben der FDA waren Stand 2025 ein aus cannabis abgeleitetes Arzneimittelprodukt und drei cannabis-bezogene Arzneimittelprodukte zugelassen. Diese Tatsache zeigt, dass cannabinoid-Pharmakologie unter definierten Bedingungen zur Medizin werden kann. Sie bedeutet nicht, dass unaufbereitete Rezeptorstimulation sicher, wirksam oder für Schizophrenie geeignet ist.
Die richtige Schlussfolgerung ist strenger und weniger beruhigend. Die CB1-Biologie ist für Psychosen relevant, weil der Rezeptor tief in Hirnschaltkreisen verankert ist, die Salienz, Kognition und Inhibition prägen. Diese Relevanz spricht für sorgfältiges Wirkstoffdesign, nicht für beiläufige Extrapolation. In der Schizophrenieforschung ist Rezeptorpharmakologie zugleich eine Karte der Möglichkeiten und der Risiken.
Was die Rezeptorwissenschaft für zukünftige cannabinoid-Medikamente bedeutet
Die cannabinoid-Medizin entfernt sich von der alten Vorstellung, ein nützliches Arzneimittel müsse lediglich „CB1 treffen“ oder „CB2 treffen“. Diese Kurzform war immer unvollständig. Sie ist heute aktiv irreführend. CB1 und CB2 sind keine einfachen An-Aus-Schalter, die sauber in Gehirn versus Immunsystem aufgeteilt sind. Sie sind Rezeptoren mit überlappenden, aber ungleich verteilten Vorkommen über Zelltypen hinweg, dynamischen Konformationen, unterschiedlichen Kopplungspräferenzen und wechselnden Rollen über Krankheitszustände hinweg. Das ist wichtig, weil derselbe Ligand in einem Gewebe therapeutisch wirken, in einem anderen berauschend sein und in einer klinischen Studie wirkungslos bleiben kann, wenn die Krankheitsgruppe zu breit ist oder das Dosierungsschema eine Desensibilisierung antreibt.
Die öffentlichen Gesundheitsrisiken sind nicht gering. Die World Health Organization schätzte, dass im Jahr 2019 200 Millionen Menschen cannabis konsumierten, was 4 % der Weltbevölkerung im Alter von 15 bis 64 Jahren entspricht. Dennoch weist die U.S. FDA weiterhin darauf hin, dass bis 2025 nur ein aus cannabis abgeleitetes Arzneimittel und drei mit cannabis in Verbindung stehende Arzneimittel zugelassen wurden. Diese Lücke zwischen weitverbreiteter Exposition und begrenzten zugelassenen Therapeutika ist ein Grund dafür, warum Rezeptorpharmakologie so wichtig ist. Ein weiterer ist die Krankheitslast: Epilepsie betrifft weltweit etwa 50 Millionen Menschen, Schizophrenie etwa 24 Millionen, und chronische Schmerzen betreffen nahezu 1 von 5 Erwachsenen in den Vereinigten Staaten. Wenn cannabinoid-Biologie bessere Arzneimittel hervorbringen soll, wird dies nicht durch eine grobe Rezeptoraktivierung geschehen.
Die grundlegenden Arbeiten von Raphael Mechoulam, Lumír Hanuš und Allyn Howlett begründeten das moderne Feld, indem sie endocannabinoid-Signalisierung und Rezeptorpharmakologie identifizierten. Die nächste Phase ist anders. Es geht weniger darum, den Nachweis zu erbringen, dass CB1 und CB2 existieren, sondern vielmehr darum, zu lernen, wie man sie präzise steuert.
| Designkonzept | Was verbessert werden soll | Warum der Artikel es für wichtig hält |
|---|---|---|
| Subtyp-Selektivität | CB1 oder CB2 gezielter bevorzugen | Der Subtyp allein garantiert weder Wirksamkeit noch Sicherheit |
| Wirksamkeitsabstimmung | Übermäßig starke Rezeptoraktivierung vermeiden | Verschiedene aktive Zustände können Nutzen, Toleranz und unerwünschte Wirkungen verändern |
| Verzerrte Signalisierung | Einen intrazellulären Signalweg gegenüber einem anderen bevorzugen | Kann helfen, nützliche Wirkungen von Belastungen zu trennen |
| Allosterische Modulation | Rezeptorantwort auf endogene Liganden abstimmen | Könnte räumlich und zeitlich normalere Signalisierung erhalten |
| Gewebebeschränkung | Exposition auf ausgewählte Organe begrenzen oder das Eindringen ins Gehirn vermeiden | Nützlich, wenn zentrale unerwünschte Wirkungen überwiegen |
Selektivität, Wirksamkeit, allosterische Modulation und Bias
Die wichtigste Lehre der jüngeren Rezeptorforschung lautet, dass „Bindung“ nicht dasselbe ist wie „Wirkung“. Ein Ligand kann CB1 gegenüber CB2 bevorzugen, oder umgekehrt, doch selbst das ist nur der Anfang. Er kann sich auch in der Wirksamkeit unterscheiden, also darin, wie stark er aktive Rezeptorzustände stabilisiert. Er kann Gi/o-Signalisierung gegenüber beta-arrestin-Rekrutierung bevorzugen oder eine schnelle Rezeptorinternalisierung stärker fördern als eine anhaltende Signalisierung an der Zelloberfläche. Diese Unterschiede können die Resultate dramatisch verändern.
Die 2026 in Frontiers in Chemical Biology veröffentlichte Übersichtsarbeit zur Struktur des cannabinoid-Rezeptors macht diesen Punkt klar: Die Subtypselektivität zwischen CB1 und CB2 beruht auf strukturellen Unterschieden auf Rezeptorebene, die Ligandenbindung, Signalisierungswirksamkeit und Rezeptorregulation prägen. Deshalb verhalten sich chemisch ähnliche cannabinoids nicht identisch. Ein Phytocannabinoid, ein endocannabinoid wie Anandamid und ein synthetischer Ligand können alle mit derselben Rezeptorfamilie in Kontakt treten und dennoch unterschiedliche nachgeschaltete Profile erzeugen. Arzneientwicklung, die Wirksamkeit und Signalisierungs-Bias ignoriert, liegt inzwischen hinter der Evidenz zurück.
CB1 ist der klarste Fall. Er wird oft als der Rezeptor diskutiert, der für psychoaktive Effekte verantwortlich ist, was zutrifft, aber unvollständig ist. CB1 reguliert auch synaptische Übertragung, Schmerzverarbeitung, Appetit, Stresskreisläufe und kortikale Signalisierung. Der 2025 im American Journal of Psychiatry erschienene Artikel argumentiert, dass biased CB1-Signalisierung eine plausible therapeutische Strategie für Schizophrenie ist. Das ist ein bemerkenswerter Wandel im Feld. Schizophrenie betrifft weltweit ungefähr 24 Millionen Menschen, und der Artikel behandelt CB1 nicht als Rezeptor, den man grundsätzlich meiden sollte. Stattdessen wird CB1 als Ziel betrachtet, dessen pathologische und therapeutische Konsequenzen davon abhängen könnten, welche intrazellulären Signalwege ein Ligand bevorzugt. Das ist klassische GPCR-Pharmakologie, nun ernsthaft auf cannabinoid-Therapeutika angewandt.
CB2 hat eine ähnliche Neubewertung erfahren. Die 2026 in Frontiers in Behavioral Neuroscience veröffentlichte Übersichtsarbeit beschreibt „ein Update der letzten 3 Jahre“ und zeigt ein wachsendes Interesse an CB2-Signalisierung bei Erkrankungen des zentralen Nervensystems, insbesondere dort, wo neuroinflammatorische und neurodegenerative Mechanismen beteiligt sind. Das bedeutet nicht, dass CB2 ein magisches entzündungshemmendes Ziel ist. Es bedeutet, dass die alte Behauptung, CB2 sei für das Gehirn irrelevant, nicht mehr zur Literatur passt. Die Expression kann unter Basalbedingungen niedrig sein und in bestimmten Zellpopulationen oder Krankheitskontexten ansteigen. Ein auf CB2 ausgerichtetes Arzneimittel kann daher stark von Zeitpunkt, Pathologie und Gewebezustand abhängen.
Allosterische Modulation könnte zu einem der nützlichsten Wege werden, diese Unterschiede auszunutzen. Orthosterische Agonisten treiben häufig eine breite Rezeptoraktivierung voran, was das Risiko von Nebenwirkungen, Toleranz und geringer Kontextselektivität erhöht. Allosterische Modulatoren hingegen können die Reaktion von Rezeptoren auf endogene Liganden fein abstimmen, statt eine maximale Aktivierung zu erzwingen. Prinzipiell könnte dies räumliche und zeitliche Aspekte der normalen endocannabinoid-Signalisierung erhalten. Es ist eine attraktive Strategie für CB1, wo direkte Agonisten oft auf zentrale unerwünschte Wirkungen stoßen, und für CB2, wo krankheitsassoziierte Hochregulation eine kontextabhängige Modulation attraktiver machen könnte als eine konstante Stimulation. Aber „attraktiv“ ist nicht „bewiesen“. Viele allosterische Ideen, die in vitro elegant aussehen, scheitern, sobald Rezeptorreserve, Gewebehomogenität und Pharmakokinetik ins Spiel kommen.
Warum Struktur- und Netzwerkbiologie heute die Arzneientwicklung leiten
Der Schwerpunkt des Feldes hat sich von einfacher Rezeptorkartierung hin zu dynamischer Rezeptorzustandsanalyse verschoben. Hier ist Strukturbiologie entscheidend. Die 2026er Frontiers-Übersichtsarbeit und die jüngste PubMed-indexierte Studie zum dynamischen Mechanismus der Subtypselektivität stellen sich beide gegen ein Schlüssel-Schloss-Modell. Selektivität ist nicht nur eine Frage davon, dass ein Ligand besser in eine Rezeptortasche passt. Rezeptoren „atmen“. Liganden stabilisieren Konformationsensembles. Endocannabinoids können Subtyppräferenzen durch dynamische Wechselwirkungen erreichen, die verändern, wie CB1 oder CB2 zwischen inaktiven, aktiven und signalkompetenten Zuständen wechselt.
Dieser Denkwechsel hat praktische Konsequenzen. Ein Medizinalchemiker fragt nicht mehr nur: „Bindet diese Substanz an CB1 oder CB2?“ Die besseren Fragen lauten: Welche Konformationen stabilisiert sie? Fördert sie Gi/o stärker als beta-arrestin? Löst sie eine schnelle Internalisation aus? Verhält sie sich in Neuronen, Mikroglia, Hepatozyten oder peripheren Nozizeptoren unterschiedlich? Kann sie auf die Peripherie beschränkt werden? Kooperiert sie mit dem endogenen Ligandentonus, der nur bei Entzündung oder Verletzung ansteigt?
Netzwerkbiologie erweitert dies noch weiter. Eine jüngere integrierte Netzwerkanalyse, die in PubMed indexiert ist, identifizierte CB1 und CB2 als hoch einflussreiche Knoten innerhalb des endocannabinoid-Systems und verknüpfte Rezeptorsignalisierung mit breiteren Stoffwechselwegen. Das ist wichtig, weil endocannabinoid-Pharmakologie nie nur Rezeptorbiologie ist. Anandamid- und 2-AG-Synthese, -Transport und -Abbau bestimmen, wie stark Rezeptoren exponiert werden. Dasselbe gilt für Lipidstoffwechsel, Entzündungsmediatoren und die Wechselwirkung mit anderen GPCR- und Ionenkanalsystemen. Ein Ligand, der in einem rekombinanten Assay sauber erscheint, kann sich in lebendem Gewebe ganz anders verhalten, wo FAAH, MAGL, Prostaglandinwege und die lokale Rezeptordichte jeweils variieren.
Diese Systemperspektive erklärt auch, warum die klinische Translation uneinheitlich war. Einige Misserfolge gingen wahrscheinlich auf schlechte Verbindungen zurück. Andere auf zu vereinfachte Krankheitsmodelle. Wenn eine Studie Patienten einschließt, deren Symptome zwar ein gemeinsames Etikett tragen, aber kein gemeinsames endocannabinoid-Signaturmuster, kann das durchschnittliche Ergebnis enttäuschend wirken, selbst wenn eine biologisch definierte Untergruppe profitieren könnte. Künftiger Erfolg wird nicht nur von Chemie abhängen, sondern von Stratifizierung: welche Patienten, welches Krankheitsstadium, welche Gewebe, welcher Rezeptorzustand, welcher endogene Tonus.
Realistische Erwartungen an die nächste Generation von Therapien
Die nächste Generation cannabinoid-basierter Arzneimittel wird wahrscheinlich enger, selektiver und weniger romantisch sein, als die öffentliche Diskussion vermuten lässt. Das ist eine gute Nachricht. Bessere Arzneimittel entstehen oft dann, wenn ein Feld aufhört, Einheitslösungen zu erwarten.
Einige Fortschritte sind bereits sichtbar. Eine auf die Peripherie beschränkte CB1-Wirkung bleibt für Schmerz- oder Stoffwechselindikationen attraktiv, weil sie zentrale unerwünschte Wirkungen verringern soll. CB2-gerichtete Verbindungen bleiben für entzündliche, neuropathische und neuroimmune Erkrankungen plausibel, insbesondere wenn krankheitsbedingte Expression therapeutische Fenster schafft. Biased CB1-Liganden könnten sich bei neuropsychiatrischen Erkrankungen als nützlich erweisen, wenn sie gewünschte Netzwerkeffekte von unerwünschter Intoxikation, Angst oder Toleranz trennen können. Allosterische Modulatoren könnten eine subtilere Kontrolle bieten als direkte Agonisten. Enzymgerichtete Ansätze, die den endocannabinoid-Tonus indirekt verändern, können weiterhin wertvoll sein, obwohl sie eigene Risiken bergen und die Rezeptor-Komplexität nicht umgehen.
Dennoch ist Realismus entscheidend. Selektivität garantiert keinen Erfolg. Ein hochselektiver CB2-Agonist kann trotzdem scheitern, wenn CB2-Expression bei der Zielerkrankung zu variabel ist oder wenn die nachgeschaltete Signalisierung nicht der eigentliche Treiber der Symptome ist. In einem Assay gemessener Bias muss in humanem Gewebe nicht denselben Bias vorhersagen. Strukturelle Momentaufnahmen sind mächtig, aber sie bleiben Momentaufnahmen. Gewebespezifität kann einige Risiken reduzieren und andere erzeugen. Und Phytocannabinoids sollten nicht als Platzhalter für die gesamte cannabinoid-Pharmakologie betrachtet werden; endocannabinoids, pflanzliche Verbindungen und synthetische Liganden können je nach Dosis, Zeitpunkt und Signalkontext sehr unterschiedliche Rezeptorverhalten hervorrufen.
Die nüchterne Prognose lautet daher: Künftige cannabinoid-Medizin wird sich wahrscheinlich dadurch verbessern, dass sie präziser wird, nicht dadurch, dass sie breiter aktiviert. Die glaubwürdigsten Wege nach vorn kombinieren Rezeptorselektivität, Signalisierungs-Bias, allosterische Kontrolle, Gewebetargeting und bessere Patientenstratifizierung. Was bekannt ist, ist erheblich: CB1 und CB2 sind dynamische Signalknoten, der strukturelle Zustand ist wichtig, und der Krankheitskontext ist wichtig. Was ungewiss bleibt, ist, ob diese mechanistische Klarheit in reproduzierbare klinische Verbesserungen bei Schmerz, psychiatrischen Erkrankungen, Epilepsie, Neurodegeneration und entzündlichen Erkrankungen übersetzt werden kann. Das Feld bleibt medizinisch bedeutsam, weil der ungedeckte Bedarf groß ist, die Biologie real ist und das frühere vereinfachte Modell endlich einer Pharmakologie gewichen ist, die sowohl Hoffnung als auch Enttäuschung erklären kann.
Quellen
- [1]Cannabis (marijuana). WHO Fact Sheet, 2024. https://www.who.int/news-room/fact-sheets/detail/cannabis-(marijuana)
- [2]Epilepsy. WHO Fact Sheet, 2024. https://www.who.int/news-room/fact-sheets/detail/epilepsy
- [3]Schizophrenia. WHO Fact Sheet, 2022. https://www.who.int/news-room/fact-sheets/detail/schizophrenia
- [4]FDA Regulation of Cannabis and Cannabis-Derived Products, Including Cannabidiol (CBD). FDA Public Health Focus, 2025. https://www.fda.gov/news-events/public-health-focus/fda-regulation-cannabis-and-cannabis-derived-products-including-cannabidiol-cbd
- [5]The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research. National Academies Press, 2017. https://nap.nationalacademies.org/catalog/24625/the-health-effects-of-cannabis-and-cannabinoids-the-current-state