






Spis treści
- Dlaczego nauki o cannabinoidach nie można sprowadzać do CB1 i CB2
- endocannabinoid system a szerszy krajobraz celów działania cannabinoidów
- Kanały TRP: czujniki ciepła, bólu i podrażnienia, w które cannabinoidy ciągle trafiają
- PPARs: cannabinoidy jako wewnątrzkomórkowe sygnały lipidowe, a nie tylko ligandy receptorów błonowych
- GPR55, GPR18 i GPR119 oraz problem receptorów sierocych GPCR
- Sygnalizacja serotoninowa: miejsce, w którym cannabinoidy przecinają się z układami 5-HT
- Poza żądaną listą: kanały sodowe i inne niekanoniczne cele już zmieniają rozmowę o bólu
- Czym różnią się poszczególne cannabinoidy, gdy przestaje się pytać wyłącznie o CB1 i CB2
- Metody mają znaczenie: dlaczego projekt testu kształtuje to, co sądzimy o działaniu cannabinoidów
- Poziomy dowodów: od szalki komórkowej do kliniki
- Bezpieczeństwo, regulacje i dlaczego farmakologia działań poza celem ma znaczenie dla zdrowia publicznego
- Odkrywanie leków: projektowanie cannabinoidów i cząsteczek inspirowanych cannabinoidami dla celów innych niż CB1/CB2
- Powszechne nieporozumienia i nierozstrzygnięte spory
- Praktyczna interpretacja dla czytelników, klinicystów i badaczy
Dlaczego nauki o cannabinoidach nie można sprowadzać do CB1 i CB2
W skrócie farmakologia cannabinoidów bywa opisywana tak: THC działa na CB1, efekty immunologiczne przebiegają przez CB2, a wszystko inne to przypis. Takie ujęcie łatwo wyjaśnić i łatwo powtarzać. Jest ono jednak często błędne na tyle, że utrudnia poważne zrozumienie bólu, stanu zapalnego, lęku, świądu, nudności, metabolizmu i neuroprotekcji.
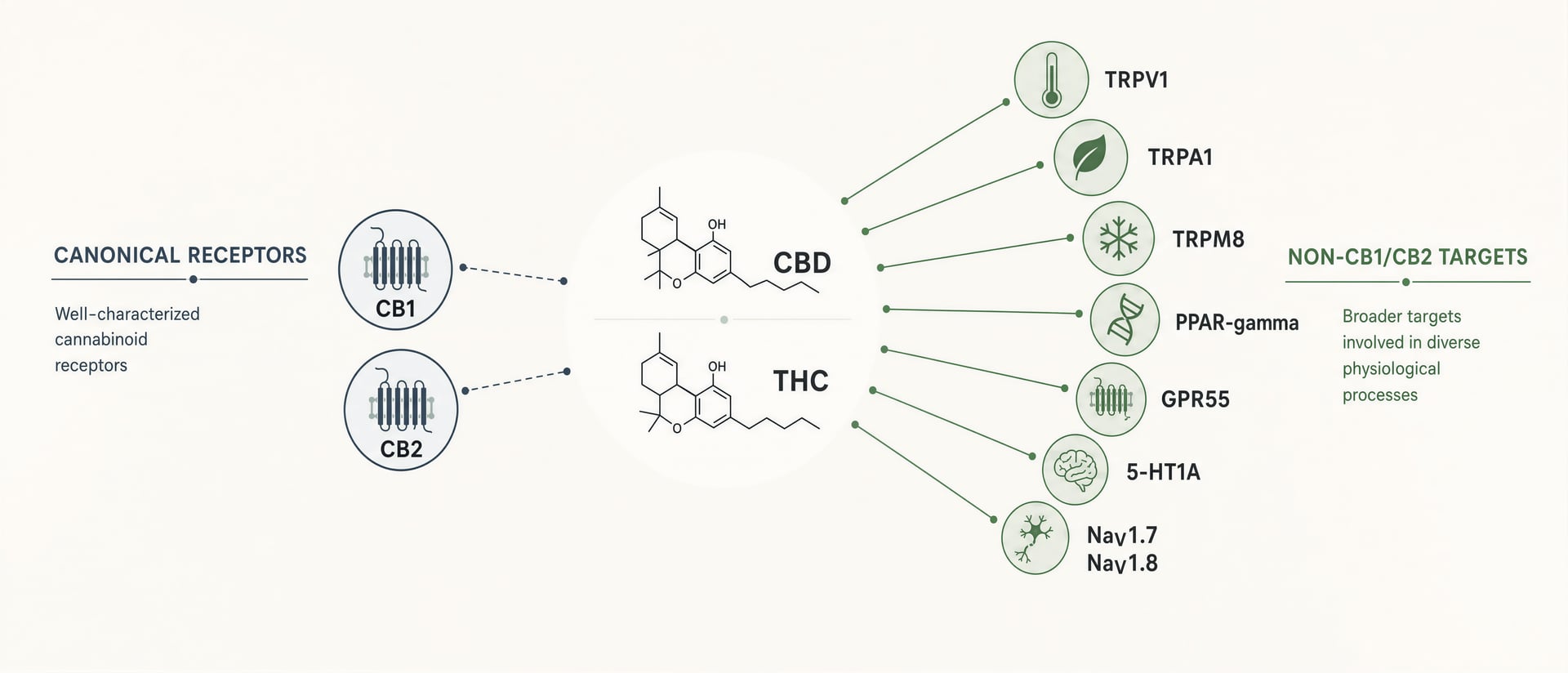
CB1 i CB2 mają znaczenie. CB1 występuje obficie w mózgu i wyjaśnia znaczną część odurzenia wywoływanego przez THC, zaburzeń pamięci, wpływu na apetyt i części jego działania przeciwbólowego. CB2 jest centralny dla wielu dyskusji o odporności i zapaleniu. Ale cannabinoidy nie są uporządkowanymi ligandami projektowanymi po jednym receptorze na każdą cząsteczkę. Są to lipofilne, elastyczne strukturalnie cząsteczki oddziałujące z szerszym polem farmakologicznym: kanałami TRP, takimi jak TRPV1 i TRPA1, receptorami jądrowymi, takimi jak PPAR-gamma, receptorami GPCR spokrewnionymi z cannabinoidami lub nadal dyskutowanymi, takimi jak GPR55 i GPR18, receptorami serotoninowymi, w tym 5-HT1A, sygnalizacją związaną z adenozyną, transportem i metabolizmem kwasów tłuszczowych oraz, w nowszych badaniach, zależnymi od napięcia kanałami sodowymi, w tym NaV1.7 i NaV1.8.[1]HHS and FDA support DEA action on dangerous 7-OH products. U.S. Department of Health and Human Services. HHS Press Room, 2025. https://www.hhs.gov/press-room/hhs-fda-support-dea-7-oh-scheduling.html
To szersze pole ma znaczenie, ponieważ mechanizm określa ryzyko i korzyść. Regulatorzy już zmagają się z tym problemem w pokrewnych sporach dotyczących polityki lekowej. W 2025 r. U.S. Department of Health and Human Services stwierdził, że „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety”, wspierając działania DEA dotyczące produktów z podwyższoną zawartością 7-OH. Sam związek nie jest cannabinoidem, ale lekcja dla polityki jest bardzo podobna: gdy chemicy zaczynają modyfikować szkielety produktów naturalnych i koncentrować metabolity, proste kategorie oparte na pochodzeniu przestają chronić społeczeństwo. Profil celów działania cząsteczki ma większe znaczenie niż to, czy publicystyka uznaje ją za znajomą.
Mit receptorów w popularnych tekstach o cannabis
Popularne omówienia cannabis zwykle przedstawiają receptory jako przełączniki włącz/wyłącz: THC aktywuje CB1, CBD „nie wiąże się silnie”, więc CBD musi być słabe albo tajemnicze. Taki opis miesza kilka odrębnych pojęć farmakologicznych w jedno nieprecyzyjne słowo: wiązać.
Klasycznym przypadkiem jest agonizm ortosteryczny. Ligand zajmuje główne miejsce aktywne receptora i stabilizuje sygnalizację. THC jest częściowym agonistą CB1 i CB2. To jeden rodzaj działania, a nie wzorzec całej biologii cannabinoidów. Związek może działać allosterycznie, zmieniając sposób, w jaki inny ligand działa na receptor, bez zajmowania tego samego miejsca. Może otwierać, uwrażliwiać lub odczulać kanał jonowy. Może wnikać do komórki i aktywować receptor jądrowy, który zmienia transkrypcję genów w ciągu godzin, a nie milisekund. Może hamować transporter, zmieniać właściwości błony albo spowalniać enzym rozkładający endogenny sygnał lipidowy.[2]EPIDIOLEX (cannabidiol) oral solution label. U.S. Food and Drug Administration. FDA drug label, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/210365s016lbl.pdf
CBD jest najczytelniejszą odpowiedzią na redukcjonizm receptorowy. Jego zatwierdzone klinicznie zastosowanie nie opiera się na agonizmie CB1. FDA label dla roztworu doustnego cannabidiol wskazuje, że jest on przeznaczony do leczenia napadów związanych z zespołem Lennoxa-Gastauta, zespołem Dravet oraz complex tuberous sclerosis u pacjentów w wieku 1 roku i starszych. Niezależnie od pełnego mechanizmu, nie da się tego efektu wyjaśnić starą opowieścią, że istotne działanie cannabinoidów oznacza silną aktywację CB1 albo CB2. W literaturze wielokrotnie wskazywano mechanizmy kandydackie, takie jak TRPV1, sygnalizacja związana z 5-HT1A, modulacja adenozyny, wpływ na wapń wewnątrzkomórkowy oraz interakcje z enzymami i transporterami. Żadnego z nich nie można uznać za jedyną odpowiedź, ale razem pokazują, dlaczego prosty mit receptorowy zawodzi.
Na tę samą stronę wskazuje także historia. Prace Raphaela Mechoulama nad endocannabinoidami otworzyły dziedzinę skoncentrowaną na anandamidzie i 2-AG, jednak nawet anandamid nie jest wyłącznie ligandem CB1. Aktywuje także TRPV1, receptor ciepła i kapsaicyny, którego szersze znaczenie dla układu czuciowego zostało uhonorowane w Nagrodzie Nobla w 2021 r. dla Davida Juliusa i Ardem Patapoutiana za odkrycie receptorów temperatury i dotyku. Gdy endogenny cannabinoid może sygnalizować zarówno przez GPCR, jak i kanał TRP, model „tylko CB1/CB2” przestaje być modelem. Staje się karykaturą.
Polypharmacology: jeden ligand, wiele celów
Lepszym punktem wyjścia jest polypharmacology. Jeden ligand, wiele celów, z różnymi powinowactwami, skutecznością, tkankami i konsekwencjami. W farmakologii słowo „brudny” bywa używane pejoratywnie, ale w przypadku cannabinoidów często jest po prostu opisowe.
Warto rozważyć, ile typów działania mieści się pod tym samym parasolem pojęciowym. THC jest częściowym agonistą CB1/CB2, ale prace z 2025 r., nagłośnione przez Hebrew University, wskazały, że THC hamuje obwodowe nocyceptory poprzez oddziaływanie z kanałami sodowymi nocyceptywnymi NaV1.7 i NaV1.8. To nie jest w ogóle agonizm receptorowy. To hamowanie kanałów jonowych w celach już uznawanych za kluczowe dla leków przeciwbólowych. Jeżeli ten kierunek potwierdzi się między gatunkami i w odpowiednich warunkach dawkowania, część działania przeciwbólowego THC może wynikać z mechanizmu bardziej przypominającego lokalny hamulec pobudliwości niż klasyczny efekt na receptorach cannabinoidowych.
CBD pokazuje inny styl nieswoistości. W różnych układach testowych raportowano wpływ na TRPV1, TRPA1, TRPM8, 5-HT1A, PPAR-gamma, GPR55 oraz ton adenozynowy, między innymi. Problemem nie jest brak mechanizmów. Problemem jest ustalenie, które z nich mają znaczenie przy stężeniach osiąganych klinicznie u ludzi. Zaangażowanie celu in vitro jest tanie. Przekład na praktykę jest trudny. Efekt mikromolarny w komórkach nadekspresjonujących receptor nie wyjaśnia automatycznie wyników u pacjenta po podaniu doustnym, po efekcie pierwszego przejścia, wiązaniu z białkami i dystrybucji tkankowej.
Inne phytocannabinoids jeszcze bardziej komplikują obraz. CBG bywa opisywany w niektórych układach jako związek oddziałujący z receptorami alfa-2 adrenergicznymi, TRP i 5-HT1A. CBC łączono z kanałami TRPA1 i TRPV. THCV może zachowywać się inaczej niż delta-9-THC w CB1 zależnie od dawki i kontekstu, a równocześnie wnosić możliwości inne niż CB1. Kwaśne cannabinoidy, takie jak CBDA i THCA, rodzą kolejne pytania, ponieważ dekarboksylacja, stabilność i tworzenie metabolitów zmieniają ekspozycję na cele. Ta sama etykieta na butelce może więc ukrywać bardzo różną farmakologię, gdy do równania wchodzą droga podania, ogrzewanie, metabolizm i formulacja.[3]Library Docking for Cannabinoid-2 Receptor Ligands. American Chemical Society. Journal of Medicinal Chemistry, 2016. https://pubs.acs.org/doi/10.1021/acs.jmedchem.6c00835
Także w obrębie farmakologii GPCR pole poszło dalej niż proste etykiety. GPR55 bywa nadal nazywany kandydatem na „CB3”, ale z dobrych powodów pozostaje to sporne; sygnalizacja, zestaw ligandów i rola fizjologiczna nie układają się gładko w klasyczny obraz receptorów cannabinoidowych. GPR18 i GPR119 są także dyskutowane w literaturze związanej z cannabinoidami, szczególnie w obszarze zapalenia, metabolizmu i sygnalizacji jelitowej, ale dowody są nierówne. Medyczni chemicy dobrze to rozumieją. Artykuł z Journal of Medicinal Chemistry z 2016 r., „Library Docking for Cannabinoid-2 Receptor Ligands”, dobrze oddaje podejście oparte na strukturze, które stanowi niemal przeciwieństwo popularnej mitologii receptorowej: projektowanie selektywne wobec celu, docking, optymalizacja szkieletu i celowe oddzielanie pożądanych efektów od niepożądanych. Pole nie pyta już „czy związek trafia w receptory cannabinoidowe?”. Pyta, które cele, w jakim stanie, w jakiej tkance, przy jakim stężeniu i z jakim biasem.
Dlaczego cele inne niż CB1/CB2 mają znaczenie kliniczne
To moment, w którym nauka przestaje być semantyczna i zaczyna wpływać na medycynę.
W bólu cele inne niż CB1 mogą być najbardziej prawdopodobną drogą do użytecznych leków o mniejszym odurzającym działaniu. TRPV1, TRPA1, obwodowe kanały sodowe i szlaki transkrypcyjne związane ze stanem zapalnym oferują sposoby ograniczania odpalania nocyceptorów lub uwrażliwienia neuroimmunologicznego bez silnej aktywacji centralnego CB1. Raport ScienceDaily z 2026 r. o związku cannabis, który „relieves pain without the high”, to sygnał na etapie badawczym, a nie gotowa odpowiedź kliniczna, ale kierunek ma sens. Jeśli można przesunąć analgezję ku obwodowym kanałom jonowym albo ograniczyć ekspozycję tkankową, dawny kompromis między ulgą w bólu a obciążeniem psychoaktywnym może się osłabić.
W stanach zapalnych i metabolizmie dobrym przykładem jest PPAR-gamma, pokazujący, dlaczego klasy receptorowe mają znaczenie. PPARs są receptorami jądrowymi, a nie błonowymi receptorami cannabinoidowymi. Ich aktywacja zmienia programy ekspresji genów związane z gospodarką lipidową, wrażliwością na insulinę i tonem zapalnym. Część efektów cannabinoidów opisywanych w modelach metabolicznych lub zapalnych lepiej pasuje do tej wolniejszej biologii transkrypcyjnej niż do szybkiej sygnalizacji CB1. Znowu jednak liczy się stężenie i dostęp wewnątrzkomórkowy. Artykuł pokazujący aktywację PPAR w teście reporterowym nie dowodzi klinicznie istotnego działania przeciwzapalnego u ludzi.[4]MIRA Pharmaceuticals Reports New Preclinical Data Demonstrating MIRA-55's Differentiated Mechanism of Action and Anxiolytic Activity Relative to THC. MIRA Pharmaceuticals. Nasdaq press release, 2025. https://www.nasdaq.com/press-release/mira-pharmaceuticals-reports-new-preclinical-data-demonstrating-mira-55s
W lęku i nudnościach stale powracają mechanizmy związane z serotoniną, szczególnie 5-HT1A. Dane są mieszane i często pośrednie, ale uporczywość tego sygnału jest znacząca. Uspokajający profil CBD trudno zamknąć wyłącznie w CB1/CB2. To jeden z powodów, dla których firmy próbują projektować odróżnialne cząsteczki inspirowane cannabinoidami, zamiast po prostu tworzyć mocniejsze analogi THC. W 2025 r. MIRA Pharmaceuticals podała, że ich kandydat MIRA-55 wykazał „differentiated mechanism of action” oraz „anxiolytic activity relative to THC”. Komunikaty prasowe firm nie są dowodem wysokiej jakości i należy traktować je właśnie tak. Mimo to pokazują, dokąd zmierza rozwój leków: od idei, że najlepszy lek cannabinoidowy to po prostu czystsza stymulacja CB1, ku zróżnicowanym mechanizmom.
Świąd, migrena, epilepsja, zaburzenia jelitowe i neuroprotekcja leżą w tej samej przestrzeni mechanistycznej. Kanały TRP regulują wzmocnienie czuciowe. GPR-y mogą kształtować sygnalizację immunologiczną i nabłonkową. PPARs zmieniają programy zapalne. Kanały sodowe bezpośrednio kontrolują pobudliwość. Szlaki serotoninowe wpływają na lęk, wymioty i reakcje stresowe. Gdy te układy zestawi się obok CB1 i CB2, a nie pod nimi, wiele rzeczywistych efektów cannabinoidów przestaje wyglądać tajemniczo, a zaczyna przypominać zwykłą farmakologię.
Uproszczony model przetrwał, bo jest łatwy. Lepszy model przetrwa konfrontację z danymi.
endocannabinoid system a szerszy krajobraz celów działania cannabinoidów
Popularne teksty o cannabis często traktują farmakologię jako opowieść o dwóch receptorach: CB1 wyjaśnia efekty psychoaktywne, CB2 wyjaśnia efekty immunologiczne, a wszystko inne to szczegół. To ujęcie jest zbyt wąskie wobec dowodów. Pomija, dlaczego cannabidiol nie daje się jasno wyjaśnić przez CB1 lub CB2, dlaczego niektóre cannabinoidy wywołują pieczenie lub analgezję przez kanały TRP, dlaczego receptory jądrowe, takie jak PPAR-γ, stale pojawiają się w badaniach stanu zapalnego, i dlaczego nawet sam THC może wpływać na kanały sodowe związane z bólem poza klasyczną sygnalizacją cannabinoidową. Jeśli dziedzina chce wyjaśniać ból, lęk, stan zapalny, kontrolę napadów lub problemy bezpieczeństwa nowych związków odurzających, redukcjonizm receptorowy musi ustąpić.
Moment regulacyjny pokazuje to wyraźnie. W 2025 r. HHS stwierdził, że „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety”, wspierając działania klasyfikacyjne wobec wzmacnianych produktów 7-OH. To stwierdzenie nie dotyczyło cannabis, ale oddaje tę samą lekcję farmakologiczną: gdy producenci przechodzą od znanych składników roślinnych do wzbogaconych, półsyntetycznych lub strukturalnie zmodyfikowanych związków odurzających, proste etykiety kategorii przestają być użyteczne. „THC-like” mówi znacznie mniej niż profil celu działania, siła, metabolity, dystrybucja tkankowa i aktywność poza celem.
Cele kanoniczne: CB1, CB2, anandamid i 2-AG
Kanoniczny endocannabinoid system nadal ma znaczenie. CB1 i CB2 są receptorami sprzężonymi z białkiem G, głównie sprzężonymi z Gi/o, zidentyfikowanymi pod koniec XX wieku i szczegółowo opisanymi przez badaczy, w tym Kena Mackiego i Vincenzo Di Marzo. CB1 występuje bardzo silnie w centralnym układzie nerwowym, szczególnie w korze, hipokampie, jądrze podstawy i móżdżku, dlatego częściowy agonizm THC w tym miejscu łączy się z odurzeniem, efektami pamięciowymi, zmianą kontroli ruchu i wpływem na apetyt. CB2 jest wzbogacony w komórkach odpornościowych i tkankach obwodowych, choć nie jest nieobecny w mózgu. Aktywacja któregokolwiek z tych receptorów zwykle zmniejsza powstawanie cAMP, moduluje kanały jonowe i zmienia uwalnianie neurotransmiterów.
Endogenne ligandy to anandamid i 2-arachidonoylglycerol, zwykle skracane do anandamidu i 2-AG. Zespół Raphaela Mechoulama był kluczowy w tej historii: anandamid zidentyfikowano w 1992 r., 2-AG wkrótce potem. Nie są one magazynowane w pęcherzykach synaptycznych jak klasyczne neurotransmitery. Powstają „na żądanie” z lipidowych prekursorów błonowych i często działają retrogradowo, przemieszczając się z komórek postsynaptycznych z powrotem do zakończeń presynaptycznych, by tłumić uwalnianie neurotransmiterów. Anandamid ulega degradacji głównie przez FAAH, a 2-AG przez MAGL. Ten cykl biochemiczny stanowi rdzeń endocannabinoid system.
Ale rdzeń nie jest całym szkieletem. Anandamid jest także agonistą TRPV1. CBD ma niskie bezpośrednie powinowactwo do CB1 i CB2 w porównaniu z THC, a mimo to ma wyraźne klinicznie istotne działania; zatwierdzony przez FDA roztwór doustny cannabidiol jest wskazany w napadach związanych z zespołem Lennoxa-Gastauta, zespołem Dravet oraz complex tuberous sclerosis u pacjentów w wieku 1 roku i starszych. To zatwierdzone użycie przypomina, że klinicznie istotne efekty cannabinoidów nie muszą pokrywać się z silnym agonizmem CB1.
Co liczy się jako cel działania cannabinoidu
Bardziej praktyczna definicja jest lepsza niż definicja purystyczna. Cel działania cannabinoidu to każde miejsce molekularne, w którym phytocannabinoid, endocannabinoid, metabolit lub szkielet inspirowany cannabinoidami wiąże się lub funkcjonalnie moduluje sygnalizację przy stężeniach, które mogą mieć znaczenie w komórkach, tkankach, zwierzętach lub u ludzi. Według tego standardu krajobraz szybko się rozszerza.
Kanały TRP są najczęściej omawianymi przykładami poza CB. TRPV1, TRPA1, TRPV2 i TRPM8 powracają w pracach o cannabinoidach. To nie jest margines. David Julius i Ardem Patapoutian otrzymali Nagrodę Nobla w 2021 r. w dziedzinie fizjologii lub medycyny „for their discoveries of receptors for temperature and touch”, przypominając, że kanały jonowe regulujące ciepło, zimno, podrażnienie i mechanosensację leżą bezpośrednio w szlakach bólowych. Anandamid aktywuje TRPV1. CBD, CBG, CBC i kwaśne cannabinoidy wykazywały aktywność wobec TRP in vitro, często przy zależności od stężenia i niekiedy z efektami dwufazowymi. Cannabinoid, który najpierw aktywuje TRPV1, może później go odczulać, dając paradoks początkowego podrażnienia, a następnie analgezji.
PPARs jeszcze szerzej otwierają perspektywę. PPAR-α i PPAR-γ są receptorami jądrowymi regulującymi transkrypcję związaną z metabolizmem lipidów i stanem zapalnym. Niektóre cannabinoidy i lipidy powiązane z endocannabinoid system działają tu bezpośrednio lub po wewnątrzkomórkowej kumulacji i metabolizmie. Są to wolniejsze efekty regulujące geny, a nie milisekundowa sygnalizacja CB1. Ma to znaczenie dla twierdzeń o przewlekłym zapaleniu, które często bardziej sensownie wyjaśnia sygnalizacja jądrowa niż ostra aktywność receptorów cannabinoidowych w synapsie.
Następnie mamy GPCR-y sieroce lub nadal sporne, zwłaszcza GPR55, GPR18 i GPR119. GPR55 był wielokrotnie proponowany jako kandydat „CB3”, ale etykieta jest przedwczesna. Receptor istnieje; klasyfikacja jest sporna. CBD bywa opisywany jako antagonista lub negatywny modulator GPR55 w układach eksperymentalnych, podczas gdy pewne endogenne lipidy i ligandy syntetyczne mogą go aktywować. GPR18 i GPR119 pojawiają się w zapaleniu, metabolizmie i sygnalizacji immunologicznej, ale dowody są nierówne i różnice gatunkowe mogą być znaczne.
Do tego szerszego mapowania należą także receptory serotoninowe, zwłaszcza 5-HT1A. Literatura o działaniu przeciwlękowym i przeciwwymiotnym CBD często wskazuje na 5-HT1A, choć nadal dyskutuje się, czy chodzi o bezpośredni agonizm, czy raczej o pośrednie ułatwienie sygnalizacji. To rozróżnienie ma znaczenie. Związek, który słabo wiąże receptor, ale niezawodnie zmienia zachowanie sieci poprzez mechanizmy allosteryczne lub sieciowe, nadal może mieć istotne działania in vivo. Tę samą ostrożność trzeba zachować wobec programów przedklinicznych raportowanych przez firmy: w 2025 r. MIRA Pharmaceuticals podała, że ich kandydat MIRA-55 miał „differentiated mechanism of action” i wykazywał aktywność anksjolityczną względem THC. To nie jest potwierdzenie korzyści klinicznej, ale pokazuje kierunek, w którym zmierza chemia leków — z dala od surowego naśladownictwa THC i ku farmakologii cannabinoidowej kształtowanej przez cele.[5]Psychoactive cannabinoid THC inhibits peripheral nociceptors by targeting NaV1.7 and NaV1.8 nociceptive sodium channels. Hebrew University of Jerusalem cannabinoids research portal. Research portal summary, 2025. https://cannabinoids.huji.ac.il/publications/psychoactive-cannabinoid-thc-inhibits-peripheral-nociceptors-targeting[6]A cannabis compound that relieves pain without the high. ScienceDaily. ScienceDaily, 2026. https://www.sciencedaily.com/releases/2026/06/260619033343.htm
Kanały sodowe również zasługują tutaj na miejsce. Raport Hebrew University z 2025 r. zidentyfikował hamowanie THC obwodowych nocyceptorów poprzez NaV1.7 i NaV1.8. To ważne, ponieważ NaV1.7 i NaV1.8 są podstawowymi celami bólowymi, a mechanizm leży poza CB1/CB2. To także wpisuje się w szerszy translacyjny trend. W 2026 r. ScienceDaily zwróciło uwagę na badania nad „a cannabis compound that relieves pain without the high”. Dokładny związek i perspektywy kliniczne wymagają starannej oceny, ale kierunek jest wiarygodny: analgezję można przynajmniej teoretycznie oddzielić od centralnego odurzenia poprzez celowanie w drogi obwodowe lub nie-CB1.
Stronniczość sygnalizacyjna Właściwość ligandu, w której stabilizuje on stany receptora sprzyjające jednej ścieżce dalszego przekazywania sygnału kosztem innej, na przykład sygnalizacji białka G zamiast rekrutacji beta-arrestin.
Powinowactwo, skuteczność, bias i okna stężeń
Ten szerszy maping celów ma sens tylko wtedy, gdy jasne są terminy farmakologiczne. Ki to stała powinowactwa wiązania: niższe Ki zwykle oznacza silniejsze wiązanie w teście konkurencyjnym. EC50 to stężenie wywołujące 50 procent mierzonego efektu funkcjonalnego. Nie są to pojęcia wymienne. Ligand może wiązać się silnie, a wywoływać słabą sygnalizację, albo wiązać umiarkowanie, a silnie zmieniać funkcję przez amplifikację szlaku.
Agonista aktywuje receptor. Antagonista blokuje aktywację przez inny ligand. Agonista odwrotny przesuwa receptory wykazujące aktywność konstytutywną ku niższej sygnalizacji bazowej. THC w CB1 jest zwykle opisywany jako częściowy agonista: nawet gdy zajmuje receptory, nie wywołuje pełnego efektu agonisty o wysokiej skuteczności. Pomaga to wyjaśnić, dlaczego różne cannabinoidy, a nawet różne syntetyczne ligandy CB1, mogą mieć bardzo różne pułapy fizjologiczne.
Bias sygnalizacyjny oznacza, że jeden ligand stabilizuje konformacje receptora faworyzujące jedną ścieżkę względem innej, na przykład sygnalizację białka G zamiast rekrutacji β-arrestyny. Jest to dziś standardowe myślenie w rozwoju leków, także w chemii leków cannabinoidowych; artykuł z 2016 r. w Journal of Medicinal Chemistry, „Library Docking for Cannabinoid-2 Receptor Ligands”, wpisuje się w tę tradycję projektowania opartego na celu. Desensytyzacja oznacza, że powtarzana lub długotrwała aktywacja może zmniejszać odpowiedź, co jest istotne dla kanałów TRP i samego CB1. Na końcu tkankowo swoiste zaangażowanie celu oznacza, że ten sam związek może trafiać w różne cele w mózgu, jelicie, skórze, komórkach odpornościowych lub nerwach obwodowych zależnie od stężenia, drogi podania, metabolizmu i lokalnej ekspresji białek. Dlatego nieswoistość in vitro nie przekłada się automatycznie na znaczenie kliniczne — ale też dlatego wyjaśnienia ograniczone do CB1/CB2 wciąż zawodzą.
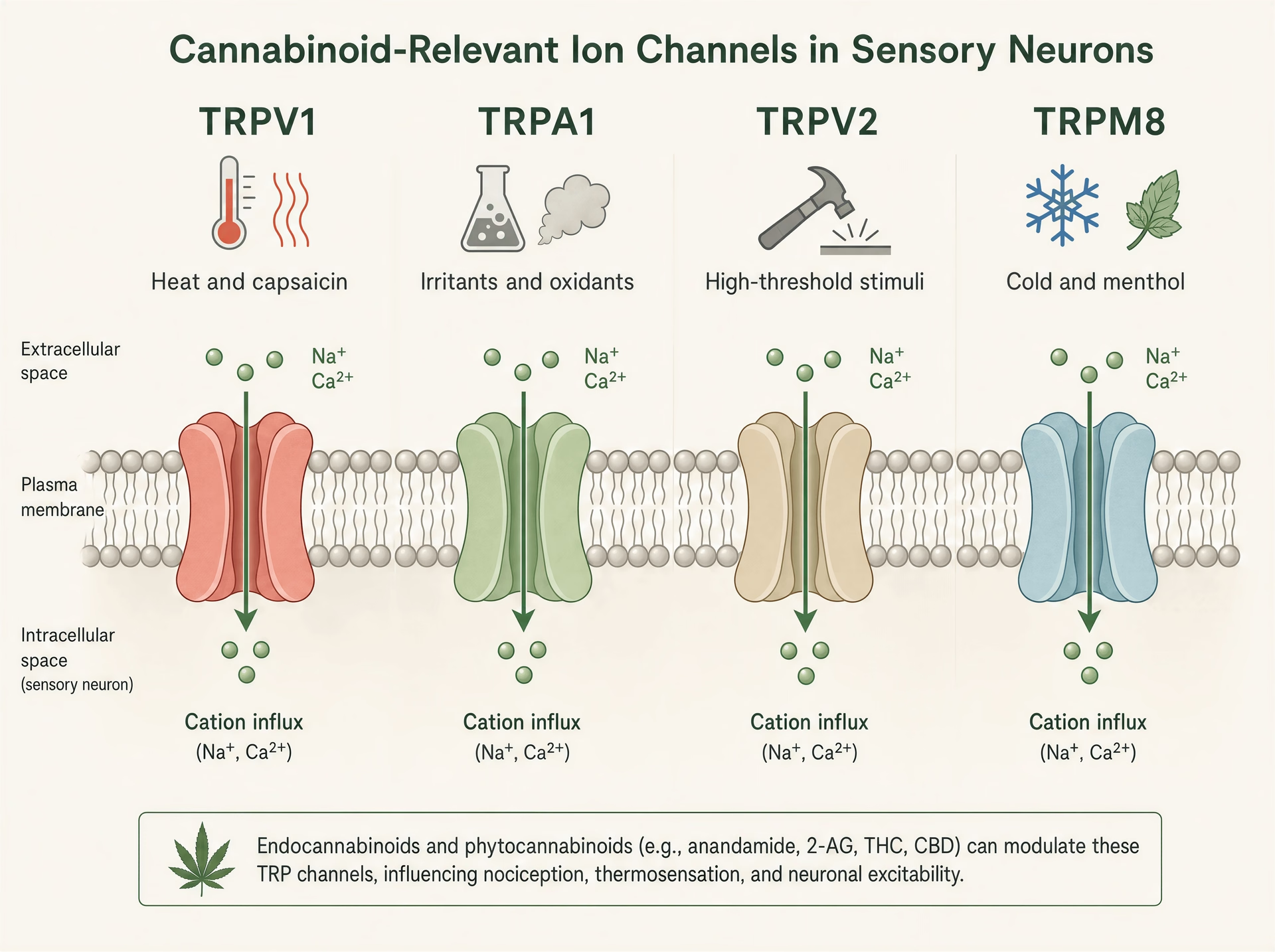
Kanały TRP: czujniki ciepła, bólu i podrażnienia, w które cannabinoidy ciągle trafiają
Zwykły skrót myślowy mówi, że cannabinoidy działają przez CB1 i CB2. To zbyt wąskie, by wyjaśnić, co wiele z tych cząsteczek naprawdę robi w tkankach. Raz po raz phytocannabinoids trafiają w kanały przejściowego potencjału receptorowego, rodzinę kanałów jonowych obecnych w nocyceptorach, keratynocytach, nerwach oddechowych, komórkach odpornościowych i innych interfejsach sensorycznych, gdzie organizm wykrywa ciepło, zimno, chemikalia, rozciąganie, uszkodzenie i stan zapalny.[7]The Nobel Prize in Physiology or Medicine 2021. The Nobel Assembly at Karolinska Institutet. Nobel Prize Press Release, 2021. https://www.nobelprize.org/prizes/medicine/2021/press-release/
Ta biologia nie jest niszowa. Była na tyle istotna dla nauki o somatosensacji, że Nagroda Nobla w 2021 r. w dziedzinie fizjologii lub medycyny trafiła do Davida Juliusa i Ardem Patapoutiana „for their discoveries of receptors for temperature and touch.” Prace Juliusa nad receptorem kapsaicyny, TRPV1, pomogły ugruntować współczesny pogląd, że sygnalizacja bólu nie jest tylko przewodem niosącym informację o uszkodzeniu; jest chemicznie bramkowana już na samym pierwszym zakończeniu czuciowym. Ma to znaczenie dla cannabinoidów, ponieważ kilka głównych cannabinoidów roślinnych oddziałuje z tym samym sprzętem molekularnym, który reaguje na papryczkę chili, olej gorczyczny, szkodliwe ciepło, czynniki chłodzące, warunki kwaśne i lipidy zapalne.
Efekt jest taki, że farmakologia wygląda chaotycznie, jeśli oczekuje się jednego receptora i jednego efektu. Ma więcej sensu, jeśli myśli się o kontroli wzmocnienia czuciowego. Wiele cannabinoidów jest słabymi do umiarkowanych ligandami receptorów CB i równocześnie bezpośrednimi modulatorami kanałów TRP. Niektóre je aktywują. Inne hamują. Jeszcze inne robią jedno i drugie zależnie od stężenia, gatunku, wariantu splicingowego, środowiska błony i tego, czy test mierzy napływ wapnia, prąd, uwalnianie neuropeptydów czy zachowanie zwierzęcia.
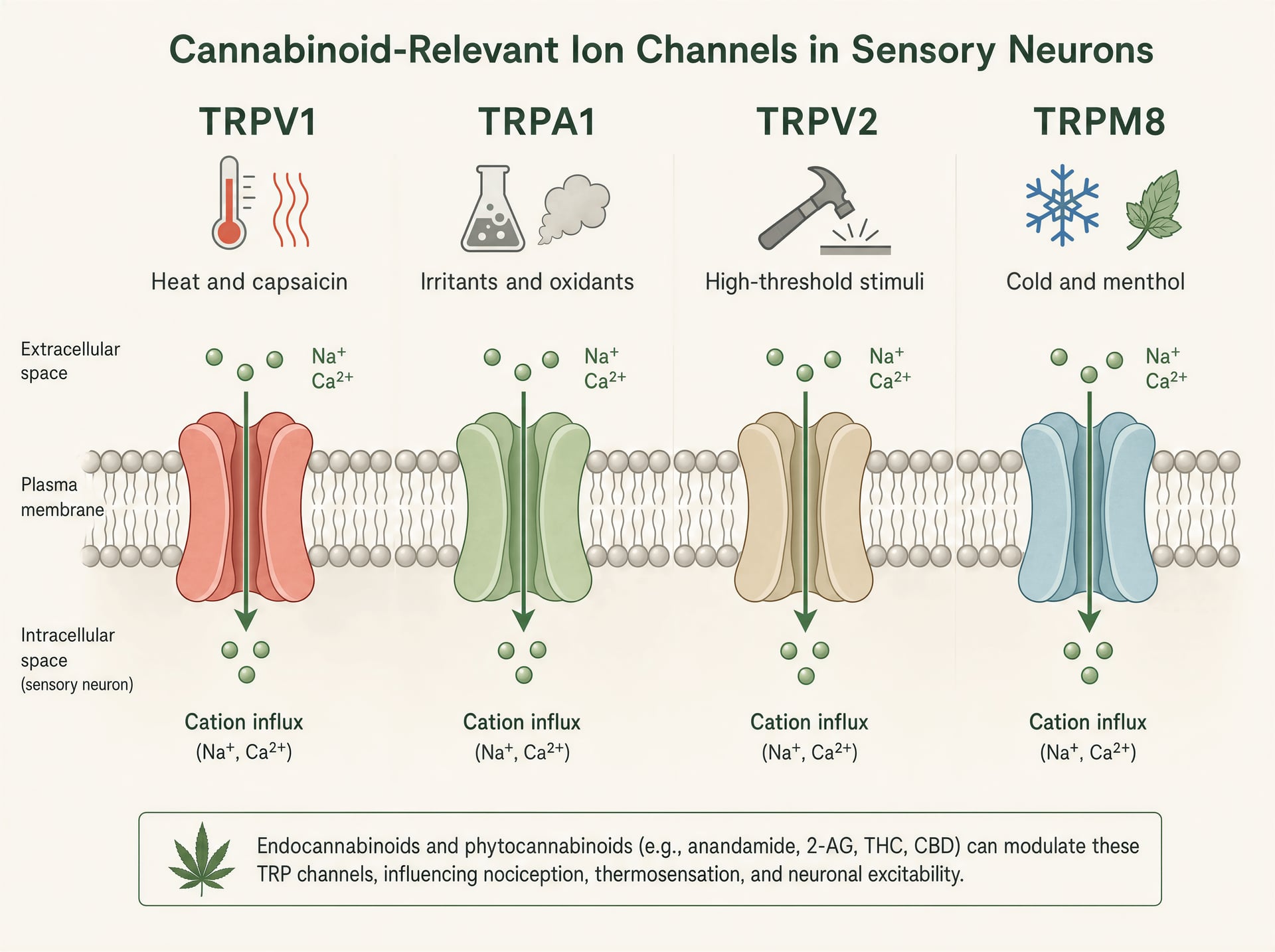
TRPV1, TRPA1, TRPV2 i TRPM8 w biologii czuciowej
Kanały TRP są detektorami wielomodalnymi. TRPV1 jest najbardziej znany: aktywowany przez kapsaicynę, szkodliwe ciepło, protony i endogenne mediatory zapalne, jest silnie ekspresjonowany w małych neuronach czuciowych odpowiedzialnych za piekący ból i zapalenie neurogenne. Otwarcie kanału powoduje napływ kationów, depolaryzację neuronu i wzrost stężenia wapnia wewnątrzkomórkowego. TRPA1 często występuje w nakładających się populacjach nocyceptorów i słynie z wykrywania drażniących elektrofilów, takich jak alliloizotiocyjanian z gorczycy i wasabi, akroleina w dymie oraz produkty stresu oksydacyjnego powstające podczas stanu zapalnego. Ma znaczenie nie tylko w bólu, ale także w świądzie, kaszlu, nadreaktywności dróg oddechowych i sygnalizacji trójdzielnej podobnej do migreny.
TRPV2 jest mniej jednoznaczny. W niektórych układach jest kanałem wysokoprogowym reagującym na temperaturę i mechanikę, ale występuje też w komórkach odpornościowych, gleju i tkankach proliferujących, dlatego stale pojawia się w dyskusjach o zapaleniu i bardziej spekulacyjnie o biologii nowotworów. TRPM8 z kolei jest klasycznym czujnikiem chłodu aktywowanym przez niską temperaturę i związki takie jak mentol i icylina. Ma również znaczenie w stanach bólowych, gdzie allodynia na zimno może być bardzo nasilona, a w niektórych kontekstach aktywność TRPM8 może tłumić ból przez przeciwstymulację na poziomie obwodowym. Ta sama rodzina, bardzo różne role sensoryczne.
To rozproszenie funkcji wyjaśnia, dlaczego efekty cannabinoidów mogą wydawać się sprzeczne. Aktywacja TRPV1 lub TRPA1 może piec. Blokowanie TRPM8 może zmniejszać odczucie chłodu, ale jednocześnie modyfikować ból na zimno. Pobudzenie TRPV2 w jednym typie komórek może wpływać na sygnalizację wapniową bez jakiegokolwiek oczywistego efektu czuciowego. Nie istnieje jeden „efekt TRP” tak samo, jak nie istnieje jeden „efekt cannabinoidu”.
CBD, CBG, CBC i THC w kanałach rodziny TRP
Spośród phytocannabinoidów CBD ma najsilniejszy i najlepiej odtwarzalny profil TRP. W heterologicznych układach ekspresyjnych CBD aktywuje ludzkie TRPV1, TRPA1 i TRPV2 przy stężeniach mikromolarnych oraz hamuje TRPM8. Często cytowane badanie De Petrocellisa i współpracowników z 2011 r., z użyciem obrazowania wapnia w transfekowanych komórkach HEK-293, wykazało, że CBD działa jako agonista TRPV1, TRPV2, TRPA1 i TRPV4, a zarazem antagonizuje TRPM8. Potencjał nie był równy: TRPA1 wykazywał szczególną wrażliwość, z aktywnością w niskich mikromolach, podczas gdy inne kanały wymagały nieco wyższych stężeń. Ten wzorzec utrzymał się na tyle dobrze, że udział kanałów TRP jest dziś częścią poważnego opisu farmakologii CBD.
CBG i CBC wpisują się w ten sam ogólny schemat, choć z własnymi cechami. CBG wielokrotnie wykazywał aktywność wobec TRPA1 i TRPV1, a także hamowanie TRPM8, co czyni go interesującym farmakologicznie w modelach bólu zapalnego i nadwrażliwości trzewnej. CBC jest słabiej zbadany niż CBD, ale dostępne prace in vitro sugerują, że także aktywuje TRPA1 i może oddziaływać na TRPV1. To nie są drobne ciekawostki z jednego testu, które nigdy nie wracają. Powtarzają się w układach rekombinowanych i w preparatach pierwotnych neuronów czuciowych, właśnie dlatego stale wracają w pracach o mechanizmach analgezji i zapalenia.
THC jest bardziej złożony. Może aktywować TRPV2, a w pewnych warunkach raportowano jego interakcję z TRPA1 i TRPV1, jednak w wielu doświadczeniach dominuje jego farmakologia związana z CB1, zwłaszcza w centralnym układzie nerwowym. Mimo to pogląd, że THC jest wyłącznie lekiem CB1, jest błędny. Najnowsze prace z Hebrew University, raportowane w 2025 r., argumentowały, że THC hamuje obwodowe nocyceptory, celując w kanały sodowe NaV1.7 i NaV1.8, czyli odrębny mechanizm nie-CB, zgodny z szerszą tezą tego tekstu: cannabinoidy często trafiają jednocześnie w wiele celów związanych z bólem. Kanały TRP są częścią tej szerszej mapy poza CB.
Potrzebna jest ostrożność. Duża część tych danych pochodzi z testów mikromolarnych, a nie każde mikromolarne stężenie w szalce odpowiada osiągalnemu stężeniu wolnemu w ludzkiej tkance docelowej. Lipofilne cannabinoidy rozdzielają się do błon, wiążą białka i tworzą metabolity; droga podania i akumulacja w tkankach mają znaczenie. Sam fakt, że FDA-approved oral cannabidiol solution jest zatwierdzony w napadach, nie dowodzi, że TRPV1 lub TRPA1 odpowiadają za jego kliniczne efekty w epilepsji. Pokazuje jedynie, że CBD wyraźnie działa u ludzi w sposób, którego nie da się ująć stwierdzeniem, że jest „nieodurzającym związkiem oddziałującym na receptory CB”. Opowieść molekularna jest większa niż taka etykieta.
Aktywność TRP jest również wrażliwa na projekt testu. Kanał może wyglądać na „aktywowany” w teście wapniowym, ponieważ równolegle zmieniają się magazyny wewnątrzkomórkowe, potencjał błonowy lub endogenne lipidy. Różnice gatunkowe mogą być realne. Tak samo zależność od stanu. Tkanka objęta zapaleniem ulega zakwaszeniu, utlenieniu i wytwarza mediatory lipidowe, które zmieniają bramkowanie TRP. Cannabinoid, który ledwie porusza kanał w warunkach bazowych, może mieć znacznie większy wpływ na uszkodzone zakończenie nerwowe.
Desensytyzacja, analgezja i dlaczego aktywacja może zmniejszać ból
To element, który dezorientuje osoby spoza specjalizacji: jeśli TRPV1 i TRPA1 są kanałami wywołującymi ból, dlaczego ich aktywacja miałaby kiedykolwiek zmniejszać ból?
Ponieważ aktywacja ostra i długotrwały efekt funkcjonalny to nie to samo.
TRPV1 jest tu klasycznym przykładem. Kapsaicyna najpierw piecze, a potem odczula nocyceptory i może dawać analgezję po powtarzanej lub wysokostężeniowej ekspozycji. Klinicznie tę zasadę wykorzystuje się w 8-procentowym plastrze kapsaicynowym w bólu neuropatycznym. Mechanizm obejmuje desensytyzację zależną od wapnia, wyczerpanie neuropeptydów takich jak substance P i CGRP, zmianę stanu fosforylacji kanału, a w niektórych przypadkach odwracalną defunkcjonalizację zakończenia nerwowego. Kanał, który na początku mocno odpala, może później stać się mniej reaktywny. Bezpośredni sygnał jest pronocyceptywny; późniejszy stan może być antynocyceptywny.
Wydaje się, że cannabinoidy wykorzystują tę samą logikę. Aktywacja TRPV1 lub TRPA1 przez CBD może wywołać napływ wapnia, a następnie zmniejszenie reaktywności kanału i tłumienie pobudliwości neuronów czuciowych. To jeden z możliwych mechanizmów, dzięki którym związek może szczypać w szalce, a jednocześnie zmniejszać hiperalgezję u zwierzęcia. Liczy się oś czasu. Liczy się też dawka. Niskie stężenia mogą uwrażliwiać lub słabo aktywować. Wyższe mogą wywoływać desensytyzację albo szersze efekty błonowe tłumiące wyładowania.
TRPA1 dodaje kolejny poziom, ponieważ jest silnie związany z drażniącymi czynnikami zapalnymi i stresem oksydacyjnym. W układach oddechowym i trójdzielnym powtarzana lub przedłużona aktywacja może zmieniać uwalnianie neuropeptydów i reaktywność odruchów. To czyni go istotnym nie tylko dla bólu, ale także dla kaszlu, migreny i stanów zaostrzenia zapalnego. Jeśli cannabinoid angażuje TRPA1, a następnie zmniejsza późniejszą reaktywność, efekt netto może być słabszym sygnalizowaniem podrażnienia, mimo że pierwszy event molekularny polegał na otwarciu kanału.
TRPM8 w wielu testach pokazuje odwrotny wzorzec niż TRPV1 i TRPA1: cannabinoidy takie jak CBD i CBG często go hamują, a nie aktywują. Może to mieć znaczenie w nadwrażliwości na zimno, gdzie nadmierna sygnalizacja TRPM8 przyczynia się do bolesnej allodynii na zimno. W tym przypadku nie ma paradoksu aktywacji prowadzącej do ulgi; prostszą hipotezą jest bezpośrednie tłumienie szlaku czuciowego na zimno. Nawet tego nie należy jednak przeceniać. W niektórych stanach bólowych aktywność TRPM8 może przeciwdziałać bólowi cieplnemu lub świądowi, więc jego blokada nie jest automatycznie korzystna.
Najsilniejsze stanowisko, jakie wspierają dowody, jest takie: kanały TRP nie są przypisem w farmakologii cannabinoidów. Są powtarzalnymi, funkcjonalnie istotnymi celami, szczególnie w obwodowych efektach czuciowych związanych z ciepłem, chemicznym podrażnieniem, bólem zapalnym, świądem i odruchami oddechowymi. Nie wyjaśniają wszystkiego. Nie zawsze są dominującym mechanizmem in vivo. Jednak każdy, kto chce zrozumieć, dlaczego CBD, CBG, CBC, a nawet THC mogą modyfikować ból i stan zapalny bez czystej zgodności z CB1 lub CB2, musi umieścić TRPV1, TRPA1, TRPV2 i TRPM8 na początku opisu, a nie na końcu.
Ma to znaczenie również dla rozwoju leków. Agencje zdrowia publicznego już rozróżniają znane cannabinoidy od chemicznie zmienionych lub wzmacnianych związków odurzających, ponieważ różnice na poziomie celu mogą zmieniać ryzyko. Ta sama zasada działa odwrotnie w terapii: jeśli można oddzielić analgezję od centralnego odurzenia, jedną z dróg jest projektowanie związków ukierunkowanych bardziej na obwodowe kanały TRP i inne cele spoza CB niż na silny, dobrze penetrujący mózg agonizm CB1. Stara opowieść oparta wyłącznie na receptorach jest zbyt mała wobec danych.
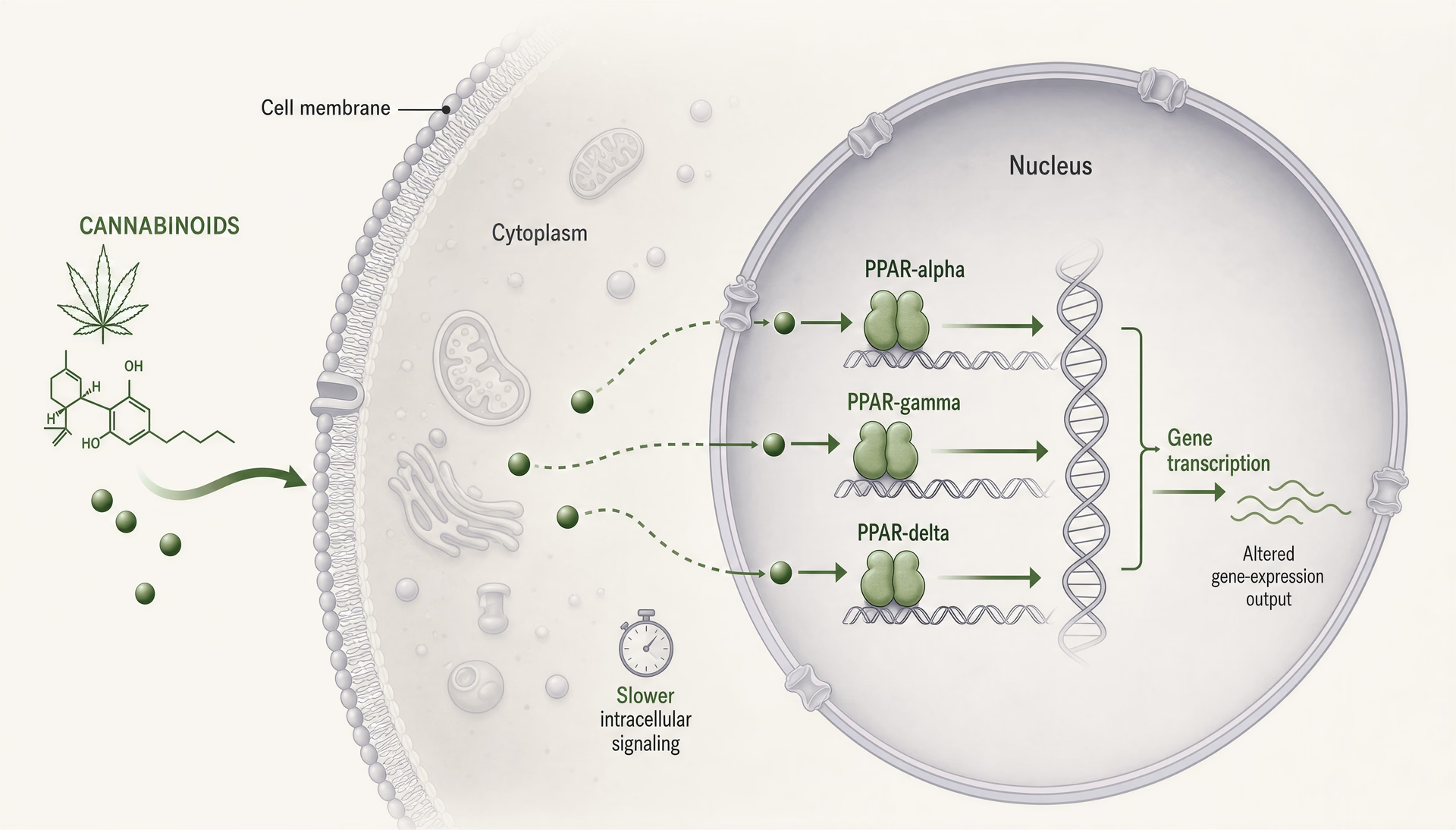
PPARs: cannabinoidy jako wewnątrzkomórkowe sygnały lipidowe, a nie tylko ligandy receptorów błonowych
Receptory aktywowane przez proliferatory peroksysomów, zwykle skracane do PPARs, zmieniają rozmowę o cannabinoidach, ponieważ znajdują się w innym miejscu i działają w innym tempie niż CB1 i CB2. CB1 i CB2 to błonowe receptory sprzężone z białkiem G, zaprojektowane do szybkiej sygnalizacji: sekundy do minut, kanały jonowe, uwalnianie neurotransmiterów, kaskady kinaz. PPARs są receptorami jądrowymi. Reagują na lipofilne cząsteczki, uruchamiają aparaturę transkrypcyjną i zmieniają, które geny komórka ekspresjonuje w ciągu godzin lub dni. Ta zmiana ma znaczenie. Oznacza, że niektóre efekty cannabinoidów mogą bardziej przypominać regulowaną lipidami reprogramację tonu zapalnego, obsługi mitochondriów, beta-oksydacji kwasów tłuszczowych, sygnalizacji włóknienia i reakcji glejowych niż klasyczny agonizm receptorowy.
To nie jest spekulacyjna rozciągłość. Cannabinoidy są silnie lipofilne, gromadzą się w błonach, rozdzielają do przedziałów wewnątrzkomórkowych i tworzą metabolity, które mogą mieć inny profil celu niż związek macierzysty. Klasa leków o takich właściwościach jest niemal stworzona do wpadania na jądrowe czujniki lipidowe. PPARs należą do najbardziej prawdopodobnych miejsc, w których tak się dzieje.
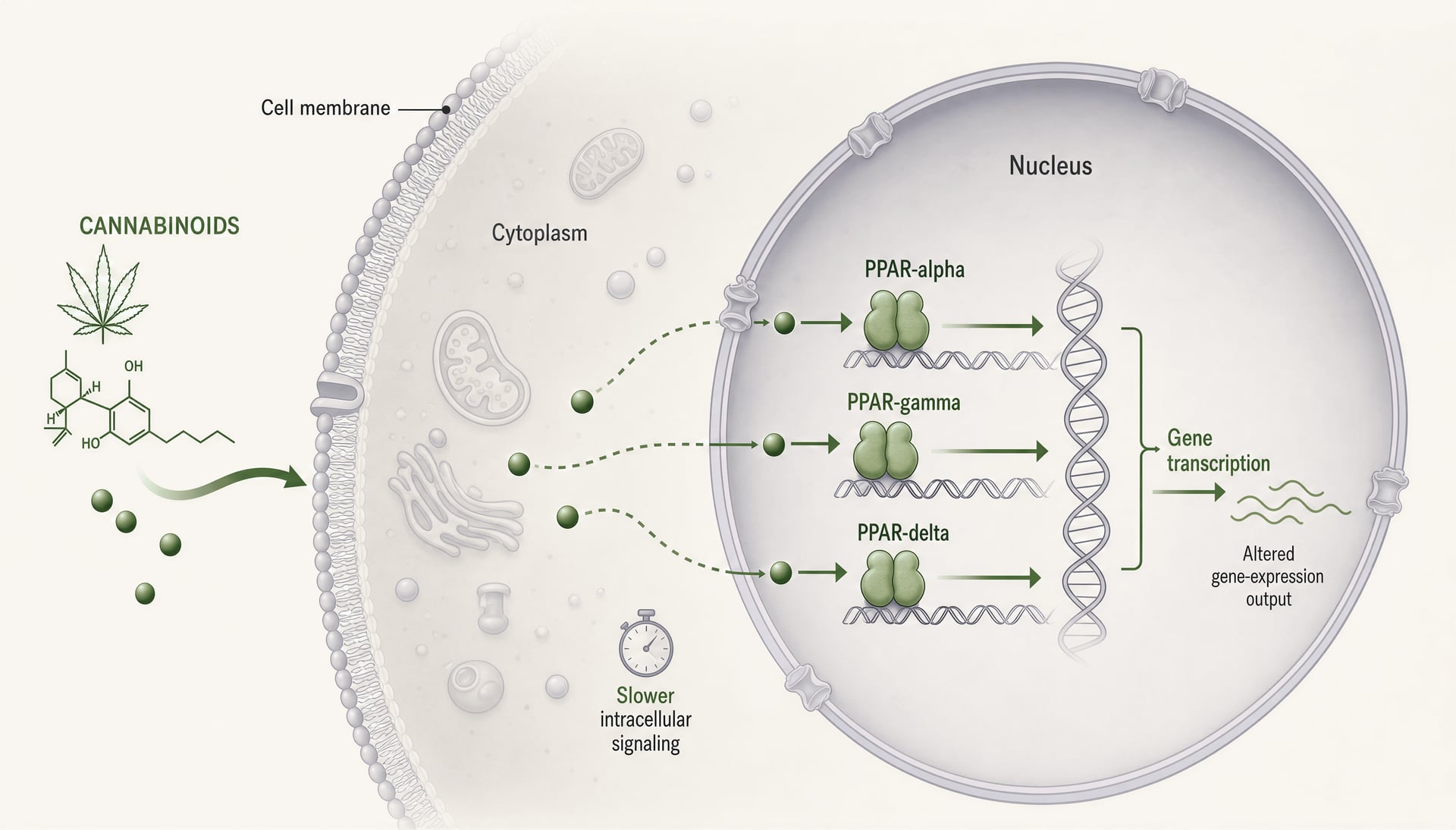
Co robią PPAR-alpha, PPAR-gamma i PPAR-delta
Trzy główne izoformy PPAR nakładają się funkcjonalnie, ale nie są wymienne. PPAR-alpha jest klasycznie związany z katabolizmem kwasów tłuszczowych. Jest obfity w wątrobie, sercu, nerkach, mięśniach i innych tkankach intensywnie spalających tłuszcz, a po aktywacji przesuwa programy transkrypcyjne w stronę beta-oksydacji, ketogenezy, gospodarki lipoproteinowej i mniejszej sygnalizacji zapalnej. Farmakolodzy znają go z leków fibratowych. W badaniach bólu i zapalenia PPAR-alpha ma znaczenie także poza metabolizmem, ponieważ może tłumić ekspresję genów zapalnych powiązaną z NF-kappaB i zmieniać sygnalizację czuciową.
PPAR-gamma jest izoformą, która ciągle pojawia się w pracach o cannabinoidach, czasem z dobrych powodów, a czasem dlatego, że to najprostsza narracja. Jest bardzo ważny dla różnicowania adipocytów i wrażliwości na insulinę, ale taki skrót spłyca obraz. PPAR-gamma reguluje polaryzację makrofagów, produkcję cytokin, odpowiedź na stres oksydacyjny, przebudowę włóknistą, zachowanie śródbłonka i aktywację gleju w centralnym układzie nerwowym. Daje to oczywistą istotność dla nieswoistego zapalenia jelit, neurozapalenia, powikłań cukrzycowych i włóknienia tkanek. Jest też celem obosiecznym: silna aktywacja może poprawiać wrażliwość na insulinę, ale wiąże się z obrzękami, przyrostem masy ciała i innymi ograniczeniami znanymi z leków tiazolidynodionowych.
PPAR-delta, znany też jako PPAR-beta/delta, otrzymuje mniej uwagi w publicznych tekstach o cannabinoidach, ale nie powinien. Jest szeroko ekspresjonowany i wspiera wykorzystanie kwasów tłuszczowych, funkcję mitochondriów, gojenie ran, biologię keratynocytów i część programów przeciwzapalnych. W zależności od kontekstu może hamować lub ułatwiać procesy chorobowe, co częściowo tłumaczy, dlaczego literatura o nim jest mniej uporządkowana. Jeśli cannabinoid lub metabolit cannabinoidu angażuje PPAR-delta, odpowiedź biologiczna może się znacznie różnić między tkankami bardziej, niż sugerowałaby prosta opowieść „agonista=korzyść”.
Mechanicznie wszystkie trzy izoformy działają jako zależne od ligandu czynniki transkrypcyjne tworzące heterodimery z retinoid X receptor i wiążące elementy odpowiedzi peroxisome proliferator response elements w DNA. Po aktywacji nie tylko włączają jeden przełącznik. Zmieniają sieci transkrypcyjne. Koaktywatory, korepresory, stan chromatyny, typ komórki, kontekst zapalny i konformacja receptora zależna od ligandu wpływają na wynik. Dwa związki mogą być nazwane agonistami PPAR-gamma, a mimo to wywoływać istotnie różną biologię.
Ta uwaga jest szczególnie ważna dla cannabinoidów, które często są cząsteczkami farmakologicznie nieswoistymi, a nie czystymi narzędziami jednego celu.
CBD i pokrewne cannabinoidy w sygnalizacji metabolicznej i zapalnej
CBD jest tu najczęściej przywoływanym przykładem, ponieważ jego profil kliniczny słabo wyjaśniają same CB1 i CB2. FDA-approved oral solution dla napadów w zespołach Lennoxa-Gastauta, Dravet oraz complex tuberous sclerosis pokazuje, że CBD jest farmakologicznie realny u ludzi, ale nie dowodzi, że jeden niecannabinoidowy cel tłumaczy wszystkie jego działania. PPAR-gamma jest jednym z najczęściej cytowanych kandydatów, ponieważ liczne badania komórkowe i zwierzęce łączyły CBD z efektami przeciwzapalnymi i metabolicznymi, które osłabiają antagoniści PPAR-gamma lub którym towarzyszą zmiany transkrypcyjne zależne od PPAR-gamma.
Szeroko cytowana praca O’Sullivana i współpracowników z 2009 r. wykazała, że CBD powoduje wazorelaksację w ludzkich tętnicach, a część efektu była wrażliwa na antagonistę PPAR-gamma GW9662, sugerując komponent zależny od PPAR-gamma. W 2011 r. Esposito i współautorzy pokazali w modelu komórkowym podobnym do choroby Alzheimera, że CBD zmniejsza neurozapalenie wywołane beta-amyloidem, a blokada PPAR-gamma osłabiała ten efekt ochronny. W 2013 r. Hind i O’Sullivan dokonali przeglądu dowodów, że cannabinoidy mogą aktywować PPARs bezpośrednio lub pośrednio, umieszczając CBD, THC, ajulemic acid, lipidy związane z anandamidem i kilka syntetycznych cannabinoidów w tym kontekście.
Wzorzec jest dość spójny, by traktować go serio: CBD często w modelach eksperymentalnych wiąże się z obniżeniem ekspresji genów zapalnych, spadkiem markerów stresu oksydacyjnego i osłabieniem odpowiedzi po antagonizmie PPAR-gamma. Traktowanie tego serio nie oznacza jednak uznania sprawy za zamkniętą. Wiele z tych badań używa mikromolarnych stężeń CBD. To ważne, ponieważ rzeczywiste wolne stężenia wewnątrz ludzkich tkanek są trudne do wywnioskowania z nominalnych stężeń w szalce. CBD wiąże się też z błonami, wpływa na gospodarkę wapnia, oddziałuje z kanałami TRP, zmienia sygnalizację adenozyny przez hamowanie transportu nukleozydów i może modyfikować ton endocannabinoid system. Każda z tych dróg może prowadzić do zmian transkrypcyjnych, które później wyglądają „jak PPAR”.
Pokrewne cannabinoidy wzmacniają argument, ale go nie upraszczają. THC w niektórych układach był raportowany jako aktywujący PPAR-gamma, choć zwykle słabiej niż klasyczne ligandy. CBDA i THCA wykazywały aktywność wobec PPAR w wybranych testach. Lipidy związane z endocannabinoid system, takie jak palmitoylethanolamide, oleoylethanolamide i niektóre oksydowane pochodne, mają silniejsze i lepiej ugruntowane relacje z PPAR-alpha i PPAR-gamma niż bardziej znane phytocannabinoids. To jeden z powodów, dla których frame oparty na wewnątrzkomórkowej sygnalizacji lipidowej jest lepszy niż wąska narracja „plant cannabinoids wiążą PPARs”. Czynna może być cząsteczka macierzysta, metabolit, współpodawany mediator lipidowy albo wtórna zmiana endogennej puli lipidów.
Ajulemic acid to użyteczne studium przypadku. Jest syntetycznym analogiem związanym z THC, ale projektowanym celowo w stronę klasycznego braku odurzenia. W badaniach przedklinicznych wykazywał działania przeciwzapalne i antyfibrotyczne, z dowodami wskazującymi na PPAR-gamma między innymi. Taka chemia leków odzwierciedla szerszy trend w dziedzinie. Już w 2016 r. artykuł ACS Journal of Medicinal Chemistry zatytułowany „Library Docking for Cannabinoid-2 Receptor Ligands” odzwierciedlał projektowanie oparte na strukturze, a nowsze programy cannabinoidowe coraz częściej dążą do oddzielenia analgezji, anksjolizy lub immunomodulacji od centralnej aktywacji CB1. Ta sama logika dotyczy szkieletów aktywnych wobec PPAR: jeśli użyteczną biologię cannabinoidową można wydobyć przez mechanizmy transkrypcyjne i obwodowe, nie ma powodu, by rozwój leków musiał pozostawać uwięziony w farmakologii podobnej do THC.
Dane o sygnalizacji metabolicznej CBD są bardziej mieszane niż dane przeciwzapalne. Niektóre badania przedkliniczne sugerują poprawę wrażliwości na insulinę, zmniejszenie zapalnych adipokin lub lepszą obsługę mitochondriów. Inne nie pokazują większej korzyści, a dane ludzkie są skąpe. Debata publiczna często wyprzedza tu dane. Sam fakt, że PPAR-gamma kontroluje glukozę i biologię tkanki tłuszczowej, nie oznacza, że CBD jest klinicznie istotnym modulatorem metabolicznym u ludzi przy standardowej ekspozycji.
Transkrypcja genów, efekty opóźnione i granice dowodów
Biologia PPAR wymusza korektę czasową. Jeśli efekt cannabinoidu pojawia się w ciągu sekund lub kilku minut, PPARs raczej nie będą głównym wyjaśnieniem. Sygnalizacja przez receptory jądrowe zwykle wymaga dostępu ligandu do przedziałów wewnątrzkomórkowych, zaangażowania receptora, zmienionego rekrutowania ko-regulatorów, zmian transkrypcyjnych, a potem konsekwencji na poziomie białek. To trwa. Godziny są możliwe. Dni są częste. Gdy prace twierdzą, że szybki efekt cannabinoidu działa „via PPAR-gamma”, uzasadniona jest ostrożność, chyba że projekt wyraźnie oddziela natychmiastową sygnalizację niegenomową od późniejszych efektów zależnych od transkrypcji.
Projekt testu pozostaje powracającym problemem. Testy reporterowe mogą wykazać, że związek zwiększa transkrypcję zależną od PPAR, ale systemy reporterowe są sztuczne i mogą przeszacowywać słabą aktywność. Badania antagonistyczne są pomocne, lecz leki takie jak GW9662 nie są magicznym serum prawdy; działania poza celem i częściowa blokada komplikują interpretację. Testy wiązania pomagają, ale samo wiązanie nie gwarantuje, że ekspozycja tkankowa osiąga potrzebne stężenie in vivo. Modele nokautu są silniejsze, choć kompensacja przez inne szlaki może zacierać obraz. Najlepsze dowody łączą metody: bezpośrednie zaangażowanie celu, farmakologię selektywną wobec receptora, zaburzenie genetyczne, odpowiednie stężenia tkankowe i przebieg czasowy zgodny z działaniem transkrypcyjnym. Duża część literatury o cannabinoidach i PPAR nie spełnia tego standardu.
Znaczenie PPAR-gamma w badaniach nad CBD jest więc jednocześnie uzasadnione i przeszacowane. Uzasadnione, ponieważ sygnał powraca w modelach naczyniowych, zapalnych, neurodegeneracyjnych i włóknieniowych. Przeszacowane, ponieważ CBD jest właśnie tego rodzaju cząsteczką lipofilną, wielocechową, dla której stężenie wewnątrzkomórkowe, aktywne metabolity i kontekst testu mogą tworzyć kuszące, ale niepełne opowieści mechanistyczne. Spadek TNF-alpha lub IL-6 po ekspozycji na CBD nie jest odciskiem palca. Jest wskazówką.
Mimo to szersza teza pozostaje prawdziwa. Cannabinoidy nie powinny być traktowane wyłącznie jako ligandy błonowych receptorów cannabinoidowych. Część z nich działa bezpośrednio lub pośrednio jako wewnątrzkomórkowe sygnały lipidowe, które mogą angażować jądrową maszynerię transkrypcyjną. Otwiera to prawdopodobne drogi do efektów przeciwzapalnych, antyfibrotycznych i neuroimmunologicznych, które są wolniejsze, mniej związane z odurzeniem i potencjalnie bardziej istotne dla długofalowej modyfikacji choroby niż ostra sygnalizacja CB1. Rodzi to również wniosek regulacyjny. Jak podkreślano w innych kontekstach, w tym w oświadczeniu HHS z 2025 r., że wzmacniane produkty 7-hydroxymitragynine stanowią „an imminent hazard to public safety”, różnice na poziomie cząsteczki mają znaczenie. Niewielkie zmiany strukturalne mogą przekierować zaangażowanie celu. W przypadku cannabinoidów i produktów do nich podobnych oznacza to, że historia bezpieczeństwa i skuteczności nie może być wyprowadzana wyłącznie ze znajomości THC, a biologia PPAR jest jednym z powodów.
GPR55, GPR18 i GPR119 oraz problem receptorów sierocych GPCR
Receptor sierocy GPCR to receptor sprzężony z białkiem G, którego endogenny ligand, rola fizjologiczna lub oba te elementy pozostają niepewne. Zdeorphanizowany receptor to taki, dla którego przekonujący endogenny aktywator został zaproponowany i odtworzony na tyle dobrze, by podtrzymywać roboczą biologię. Brzmi to porządnie. W praktyce rzadko takie jest. Farmakologia cannabinoidów ciągle wpada w ten bałagan, ponieważ endocannabinoids i phytocannabinoids są lipofilne, aktywne błonowo i nieswoiste: mogą zmieniać przepływ wapnia, aktywność kinaz czy transkrypcję w sposób wyglądający na receptorowy, nawet gdy bezpośredni cel nie jest ustalony. Tak właśnie GPR55, GPR18 i GPR119 weszły do dyskusji jako „nieklasyczne receptory cannabinoidowe”.
Pokusa stworzenia nowej etykiety receptorowej jest silna. Dobrze wygląda w nagłówkach. Wyprzedza też dowody. GPR55 był najbliżej nazwania „CB3”, lecz dziedzina nigdy nie osiągnęła spójności, która wspiera CB1 i CB2. Ta sama ostrożność dotyczy jeszcze bardziej GPR18 i GPR119.
Dlaczego GPR55 bywał kiedyś nazywany możliwym receptorem cannabinoidowym
GPR55 sklonowano w 1999 r., a wczesne badania ekspresji umieściły go w tkankach istotnych dla biologii cannabinoidów: obszarach mózgu, zwojach korzeni tylnych, śledzionie, przewodzie pokarmowym, naczyniach, komórkach odpornościowych i komórkach związanych z kością, w tym osteoklastach i komórkach linii osteoblastycznej. To miało znaczenie. Receptor obecny w szlakach bólowych, tkankach zapalnych i kości natychmiast nasuwa porównanie z CB1 i CB2, zwłaszcza gdy ligandy cannabinoidowe wydają się zmieniać jego odczyty.
Jego profil sygnalizacyjny wyglądał też wystarczająco inaczej, by zainteresować. W odróżnieniu od CB1 i CB2, które głównie sprzęgają się z Gi/o i zwykle hamują cyklazę adenylanową, GPR55 najczęściej sygnalizuje przez Gα12/13 i czasem przez ścieżki związane z Gq, aktywując RhoA, fosfolipazę C, ERK i uwalnianie wapnia wewnątrzkomórkowego. W testach komórkowych charakterystycznym odczytem jest często przejściowy wzrost wapnia. To sprawiało, że GPR55 łatwo było „zobaczyć” w układach heterologicznych, ale też łatwo przecenić, bo testy wapniowe są wrażliwe na gęstość receptora, tło komórkowe, lipofilność ligandu i czas testu.
Konkretny powód, dla którego GPR55 stał się kandydatem na receptor cannabinoidowy, był taki, że kilka cannabinoidów i ligandów podobnych do cannabinoidów dawało w nim mierzalne efekty. Ryberg i współpracownicy w British Journal of Pharmacology w 2007 r. raportowali, że GPR55 może być aktywowany przez wiele ligandów cannabinoidowych i zaproponowali go jako „a novel cannabinoid receptor.” Ten artykuł stał się historycznym punktem zwrotnym. Nie rozstrzygnął pytania; stworzył je.
Niedługo potem pojawiły się pęknięcia. Niektóre grupy uznały, że lizofosfatydyloinozytol, zwłaszcza gatunki 2-arachidonoyl LPI, jest bardziej przekonującym endogennym agonistą niż jakikolwiek klasyczny cannabinoid. Oka i współpracownicy w 2007 r. oraz późniejsze badania mocno popchnęli ten pogląd. Inni obserwowali, że związki często omawiane w badaniach nad cannabinoidami zachowują się przy GPR55 niespójnie: cannabidiol (CBD) często wyglądał jak antagonista lub negatywny modulator w niektórych testach, podczas gdy Δ9-THC był słaby, częściowy albo nieaktywny zależnie od układu. Abnormal cannabidiol, O-1602 i niektóre syntetyczne cannabinoidy czasem wykazywały wyraźniejszą aktywność niż sam THC. To nie jest obraz czystego trzeciego receptora cannabinoidowego.
Mimo to biologia GPR55 jest realna, nawet jeśli etykieta jest niestabilna. W badaniach bólu receptor jest obecny w neuronach czuciowych i obwodach rdzeniowych, a genetyczne lub farmakologiczne przerwanie sygnalizacji GPR55 zmniejszało nadwrażliwość mechaniczną w niektórych modelach gryzoni. Staton i współpracownicy w Pain (2008) powiązali aktywację GPR55 z procesami bólu zapalnego i neuropatycznego, a antagonizm zmniejszał nadwrażliwość. Efekt nie jest jednak uniwersalny między modelami i ligandami. Część danych sugeruje sygnalizację pronocyceptywną przez mobilizację wapnia i zwiększoną pobudliwość neuronalną; inne zestawy danych są słabsze lub ograniczone do modelu. Najbezpieczniej uznać, że GPR55 może uczestniczyć w sygnalizacji bólowej w niektórych kontekstach, szczególnie zapalnych, ale nie jest głównym przełącznikiem bólu.
Biologia kości daje mocniejszy sygnał. Dlaczego? Ponieważ fenotypy knockout GPR55 trudniej zrzucić na artefakty testu. W 2009 r. Whyte i współpracownicy opisali w PNAS, że myszy pozbawione GPR55 miały większą masę kostną i zaburzoną funkcję osteoklastów, co wskazywało, że GPR55 promuje resorpcję osteoklastyczną. Miało to sens mechanistyczny w kontekście sygnalizacji związanej z wapniem i RhoA. Osteoklasty zależą od przebudowy cytoszkieletu i lokalnej obsługi wapnia; GPR55 pasuje do tej maszynerii lepiej niż CB1. Jeśli cannabinoid lub związek podobny do cannabinoidu moduluje tu GPR55, konsekwencja fizjologiczna może być znaczna.
Trzecim głównym motywem jest zapalenie. GPR55 występuje w komórkach związanych z odpornością, a jego aktywację wiązano z uwalnianiem cytokin, zachowaniem leukocytów i naczyniowymi odpowiedziami zapalnymi. Ale znowu kierunek nie jest całkiem jednolity. W niektórych preparatach aktywacja GPR55 wygląda prozapalnie, w innych bardziej regulacyjnie, co prawdopodobnie odzwierciedla typ komórki, bias ligandu i cross-talk receptorowy, a nie prostą sprzeczność. Receptor sprzęgający się wieloma szlakami i znajdujący się w różnych środowiskach błonowych nie daje jednego uniwersalnego wyniku.
Ta złożoność wyjaśnia wieloletni spór o agonistę i antagonistę w literaturze cannabinoidowej. CBD jest tu najczytelniejszym przykładem. W kilku badaniach CBD często zachowywał się jak antagonista GPR55 lub inhibitor funkcjonalny, osłabiając sygnalizację wapniową wywołaną przez LPI. Lauckner i współpracownicy w 2008 r., w szeroko cytowanej pracy PNAS, pokazali, że aktywacja GPR55 zwiększała stężenie wapnia wewnątrzkomórkowego i promowała uwalnianie neurotransmiterów, podczas gdy CBD przeciwstawiał się części tej sygnalizacji. Z tego wyrosła trwała hipoteza, że niektóre efekty CBD, zwłaszcza w modelach napadów i zapalenia, mogą częściowo wynikać z blokady GPR55, a nie z działania na CB1 lub CB2. To jest hipoteza prawdopodobna. Nie jest jednak dowiedziona jako dominujący mechanizm u ludzi.
THC jest jeszcze bardziej niejednoznaczny. Niektóre doniesienia klasyfikują go jako niskopotencyjny agonista GPR55; inne znajdują znikomą skuteczność; jeszcze inne wskazują na zachowanie zależne od rezerwy receptora lub mierzonego szlaku. Ligand może wyglądać jak agonista w teście β-arrestin, być neutralny w teście wiązania i antagonistyczny w teście wapniowym, jeśli układ jest nadekspresyjny albo spolaryzowany szlakowo. To nie jest drobny szczegół techniczny. To jest sedno sprawy.
Mieszane dowody dla GPR18 i GPR119
GPR18 był często omawiany, ponieważ w niektórych układach reaguje na N-arachidonoyl glycine, lipid powiązany z endocannabinoidami, a także dlatego, że abnormal cannabidiol i podobne związki wykazywały efekty naczyniowe lub immunologiczne, które niektórzy autorzy mapowali na GPR18. Ekspresję opisywano w komórkach odpornościowych, mikrogleju, śledzionie i niektórych tkankach obwodowych. To czyniło go atrakcyjnym kandydatem dla regulacji zapalenia, migracji komórek odpornościowych i być może bólu.
Ale farmakologia od początku była nierówna. Kohno i współpracownicy w 2006 r. wspierali aktywację GPR18 przez N-arachidonoyl glycine. McHugh i współpracownicy później łączyli GPR18 z migracją mikrogleju i sygnalizacją zapalną. Potem pojawiły się problemy z replikacją. Niektóre laboratoria nie mogły odtworzyć odpowiedzi ligandowej w systemach transfekowanych. Inne znajdowały silną zależność od tagowania receptora, linii komórkowej lub ortologa gatunkowego. Receptor, który „działa” tylko w jednej architekturze testu, nie jest zdeorphanizowany w żadnym stabilnym sensie. Dla cannabinoidów dowody są tu słabsze niż sugerują popularne podsumowania. Być może istnieje tu realna biologia, ale argument za GPR18 jako prawdziwym receptorem cannabinoidowym pozostaje słaby.
GPR119 jest inny. Jest znacznie mniej prawdopodobny jako receptor cannabinoidowy, mimo że czasem pojawia się na szerokich listach receptorów „non-CB”. GPR119 wiąże się przede wszystkim z wykrywaniem lipidów w komórkach beta trzustki i komórkach enteroendokrynnych, sprzęga się przez Gs, zwiększa cAMP i promuje zależne od glukozy wydzielanie insuliny oraz incretin. Oleoylethanolamide jest lepiej ugruntowanym kandydatem na ligand endogenny niż jakikolwiek klasyczny cannabinoid. Ponieważ niektóre etanoloamidy kwasów tłuszczowych są strukturalnie bliskie chemii endocannabinoidów, GPR119 bywa wciągany do dyskusji o cannabinoidach przez asocjację. To głównie pomieszanie kategorii. Nakładanie się jest sąsiedztwem chemicznym, a nie mocnym dowodem, że THC, CBD lub główne phytocannabinoids działają istotnie przez GPR119 przy fizjologicznych stężeniach.
Co w nagłówkach robi farmakologia receptorów sierocych źle
Standardowy błąd mediów jest prosty: jeden pozytywny test sygnalizacyjny staje się „naukowcy odkryli nowy receptor cannabinoidowy.” Taki skok ignoruje co najmniej cztery filtry.
Po pierwsze, zależność od testu. Mobilizacja wapnia, rekrutacja β-arrestiny, fosforylacja ERK, dynamic mass redistribution i wiązanie radioligandu nie zadają tego samego pytania. Lipofilny ligand może zaburzać błony, zmieniać transport receptorów albo wykazywać bias szlakowy. Jeśli receptor jest nadekspresjonowany, słabe związki zaczynają wyglądać na silne.
Po drugie, różnice gatunkowe. Ludzki GPR55 nie jest pod każdym względem farmakologicznym myszy GPR55, i to samo dotyczy GPR18. Profil ligandu zbudowany w komórkach HEK293 z ludzkim receptorem nie musi przewidywać badania bólu u szczura.
Po trzecie, stężenie. Wiele prac o cannabinoidach raportuje aktywność mikromolarną in vitro. To może mieć znaczenie farmakologiczne, ale nie automatycznie. Stężenia tkankowe po inhalacji, podaniu doustnym, metabolizmie pierwszego przejścia lub lokalnej akumulacji w tłuszczu i błonach różnią się ogromnie. Wiązanie in vitro to nie mechanizm kliniczny.
Po czwarte, kontekst. Receptor w komórkach odpornościowych może pośredniczyć w jednym efekcie; ten sam receptor w osteoklastach — w innym. Dodaj cross-talk z kanałami TRP, PPARs, receptorami serotoninowymi, a nawet kanałami sodowymi, i czysta opowieść o jednym ligandzie i jednym receptorze szybko się rozpada.
Dlatego „CB3” nigdy się nie przyjął. GPR55 ma wiarygodną biologię w sygnalizacji wapniowej, bólu, przebudowie kości i zapaleniu. Ma też sprzeczną farmakologię cannabinoidową, dużą wrażliwość na projekt testu i silny konkurencyjny argument, że to lipidy z rodziny LPI są jego głównymi ligandami fizjologicznymi. GPR18 jest jeszcze bardziej niepewny. GPR119 zazwyczaj nie należy do tej samej kategorii poza przypomnieniem, że lipidowe GPCR-y łatwo błędnie łączyć z cannabinoidami.
Dla nauki o cannabinoidach wniosek jest prosty: powściągliwość. Te receptory mogą mieć duże znaczenie. Po prostu nie uzasadniają przedwczesnego przemianowania.
Sygnalizacja serotoninowa: miejsce, w którym cannabinoidy przecinają się z układami 5-HT
Serotonina to miejsce, w którym wiele popularnych twierdzeń o CBD staje się zarówno bardziej prawdopodobnych, jak i bardziej śliskich. Część prawdopodobna jest prosta: w testach komórkowych, modelach lęku u gryzoni, paradygmatach stresu i w niewielkiej liczbie badań eksperymentalnych u ludzi 5-HT1A wciąż pojawia się jako istotny węzeł efektów behawioralnych CBD. Część śliska polega na tym, że „działa na serotoninę” może znaczyć kilka różnych rzeczy. Może oznaczać bezpośredni agonizm w miejscu ortosterycznym. Może oznaczać pozytywną modulację allosteryczną. Może oznaczać ułatwienie sygnalizacji receptora bez wysokiego powinowactwa wiązania. Albo może oznaczać, że CBD zmienia aktywność sieciową powyżej lub poniżej neuronów serotoninergicznych, dając wynik zależny od serotoniny, ale nie będąc klasycznym lekiem serotoninowym.
To rozróżnienie ma ogromne znaczenie. Jeśli związek uspokaja zachowanie w sposób blokowany przez antagonistę 5-HT1A, takiego jak WAY-100635, nie dowodzi to samo w sobie, że związek jest agonistą 5-HT1A. Dowodzi zależności od sygnalizacji 5-HT1A w danym modelu. To nie jest to samo, a przekazy o cannabinoidach często te rzeczy mieszają.
5-HT1A i pytanie o lęk
Najmocniejszym łącznikiem serotoninowym dla cannabinoidów, zwłaszcza CBD, jest 5-HT1A. Jest to receptor serotoninowy sprzężony z Gi/o, obecny zarówno jako autoreceptor na neuronach serotoninergicznych jądra szwu, jak i jako receptor postsynaptyczny w regionach istotnych dla lęku, w tym w hipokampie, ciele migdałowatym i korze przedczołowej. Leki aktywujące lub wykorzystujące ten układ mogą w niektórych sytuacjach zmniejszać lęk, ale liczy się lokalizacja receptora: hamowanie wyładowań serotoninergicznych przez autoreceptory nie jest tym samym, co kształtowanie sygnalizacji postsynaptycznej w obwodach limbicznych.
CBD wszedł do tej dyskusji przez prace przedkliniczne z lat 2000. i 2010., pokazujące efekty podobne do anksjolitycznych w testach takich jak elevated plus maze, Vogel conflict test i contextual fear paradigms, częściowo blokowane przez WAY-100635. Jednym z często cytowanych artykułów jest Campos i Guimarães, 2008, które wykazały, że CBD podawany do prelimbic reduced restraint-stress-related cardiovascular responses, a mechanizmy 5-HT1A częściowo uczestniczyły w efekcie. Innym ważnym badaniem u ludzi jest Bergamaschi i wsp., 2011: w teście symulowanego wystąpienia publicznego 600 mg CBD doustnie zmniejszało lęk u osób z social anxiety disorder względem placebo. Artykuł ten nie dowiódł mediacji przez 5-HT1A u ludzi, ale pasował do wzorca przedklinicznego i uczynił serotoninę poważnym kandydatem mechanistycznym, a nie sloganem marketingowym.
Farmakologia receptorowa nigdy jednak nie sprowadziła się do prostego „CBD jest agonistą serotoniny”. Wczesne badania in vitro sugerowały, że CBD może wypierać ligandy z ludzkich receptorów 5-HT1A i działać jako agonista w niektórych testach sygnalizacyjnych, ale powinowactwo było umiarkowane i zależne od układu. Russo i współpracownicy w 2005 r. opisali CBD jako agonistę sklonowanych ludzkich receptorów 5-HT1A w testach wiązania [35S]GTPγS. To było wpływowe, ale późniejsze badania skomplikowały obraz. Niektóre grupy widziały słabą aktywność bezpośrednią. Inne obserwowały funkcjonalne wzmocnienie lepiej wyjaśniane przez efekt allosteryczny lub błonowy. Literatura jest zgodna tylko w jednym punkcie: 5-HT1A ma większe znaczenie dla farmakologii CBD związanej z lękiem niż same CB1 czy CB2.
Dlatego redukcjonizm receptorowy zawodzi. Gdyby CBD był po prostu czystym agonistą 5-HT1A, jego profil powinien przypominać znane leki serotoninergiczne znacznie bardziej niż przypomina. Tymczasem sygnał behawioralny jest silnie zależny od kontekstu, często pokazując odwrócone krzywe dawka-odpowiedź typu U. W niektórych testach u gryzoni dawki umiarkowane zmniejszają zachowanie podobne do lęku, podczas gdy niższe lub wyższe dają mniej. To sygnał ostrzegawczy przeciw opowieści o jednym receptorze. Aktywacja TRPV1 przy wyższych stężeniach jest jedną z proponowanych przyczyn. Podobnie jak wpływ na ton endocannabinoid system, wychwyt adenozyny i gospodarkę wapniową wewnątrzkomórkową. Cząsteczka może angażować 5-HT1A i nadal nie zachowywać się jak podręcznikowy lek 5-HT1A.
Bezpośrednie wiązanie a pośrednie efekty serotoninowe
Najlepszy sposób czytania dowodów serotoninowych to podział na poziomy. Na poziomie molekularnym istnieją dowody na bezpośrednią interakcję CBD z 5-HT1A, ale nie w tym czystym, wysokopowinowactwowym i wysokoskutecznym sensie, który zamyka sprawę. W zależności od układu testowego CBD bywał opisywany jako słaby agonista, częściowy agonista lub pozytywny modulator allosteryczny. Spór nie jest czystą semantyką. Agoniści ortosteryczni zajmują główne miejsce wiązania serotoniny. Pozytywni modulatory allosteryczni zmieniają zachowanie receptora z innego miejsca i mogą wzmacniać odpowiedzi na endogenną serotoninę bez silnej własnej aktywacji receptora. Te mechanizmy mają różne implikacje dla dawki, czasu działania, działań niepożądanych i translacji do ludzi.
Dane sygnalizacyjne komórkowe często wskazują bardziej na ułatwienie niż na brutalną aktywację. W niektórych preparatach CBD wzmacnia kaskady sygnalizacyjne 5-HT1A, w tym efekty na ERK i inne szlaki downstream, bardziej niż wynikałoby to z jego słabego wiązania. Możliwych wyjaśnień jest kilka. CBD jest silnie lipofilne i rozdziela się do błon, gdzie może zmieniać mikrośrodowisko receptora i sprzęganie z białkiem G. Może też pośrednio podnosić sygnalizację anandamidu, a cross-talk endocannabinoid-serotonin w grzbietowym jądrze szwu i przodomózgowiu jest dobrze udokumentowany. Do tego dochodzi adenozyna: CBD hamuje aktywność transporterów nukleozydów w niektórych układach, zwiększając pozakomórkową adenozynę i zmieniając pobudliwość neuronów w sposób wpływający na obwody serotoninowe. Nic z tego nie czyni 5-HT1A nieistotnym. Czynią go osadzonym w sieci.
Farmakologia zwierzęca daje mocniejsze dowody zależności od serotoniny niż od bezpośredniego agonizmu. Raz po raz WAY-100635 osłabia działanie CBD w modelach lęku, paniki, nudności i stresu. Resstel i wsp., 2009, wiązali na przykład osłabienie ostrych odpowiedzi na restrykcyjny stres przez CBD z mechanizmami 5-HT1A. Prace Rock i Parker nad nudnościami i nudnościami antycypacyjnymi u gryzoni również wskazywały 5-HT1A jako element profilu przeciwwymiotnego CBD. To użyteczne wyniki, ale należy je czytać jako dowody na zaangażowanie szlaku. Jeśli zablokowanie 5-HT1A usuwa efekt, szlak jest w to zaangażowany. Nie rozstrzyga to, czy receptor jest wiązany bezpośrednio, modulowany allosterycznie, czy rekrutowany przez zmiany sieciowe.
Dowody u ludzi pozostają skromne. Badanie Bergamaschi z 2011 r. często się cytuje, ponieważ pokazało mierzalny sygnał anksjolityczny w social anxiety podczas przemawiania publicznego. Mniejsze badania obrazowe wykazały, że CBD zmienia aktywację układów limbicznych i paralimbicznych podczas zadań emocjonalnych. Żadne z tych badań nie określiło jednak zajęcia receptorów 5-HT1A u ludzi tak, jak robią to badania PET dla uznanych leków serotoninowych. Ta nieobecność jest ważna. Wyprowadzamy mechanizm z zbieżności danych, a nie mierzymy go bezpośrednio przy dawkach klinicznych.
Uspokajające efekty CBD zależą częściowo od sygnalizacji 5-HT1A.Limited evidence
Dlaczego uspokajający profil CBD opiera się prostym etykietom receptorowym
CBD ma już jedno zatwierdzone zastosowanie FDA i nie jest nim lęk. Etykieta FDA z 2024 r. dla roztworu doustnego cannabidiol ogranicza wskazanie do napadów związanych z Lennox-Gastaut syndrome, Dravet syndrome lub complex tuberous sclerosis u pacjentów w wieku 1 roku i starszych. To dobry test przeciwko przesadzie. Cząsteczka może dawać wiarygodne sygnały anksjolityczne, nie mając jeszcze ustalonej skuteczności w lęku na poziomie regulacyjnym, i może uczestniczyć w sygnalizacji serotoninowej bez jasnego przynależenia do kategorii leków serotoninowych.
Część problemu dotyczy skali. In vitro cannabinoids są farmakologicznie chaotyczne. In vivo są jeszcze bardziej chaotyczne, ponieważ dystrybucja, metabolizm, akumulacja tkankowa i różnice gatunkowe zmieniają to, które cele są istotne. Efekt receptora widoczny przy 10 mikromolach w transfekowanych komórkach może być nieistotny po zwykłym podaniu doustnym, podczas gdy słabszy z pozoru efekt in vitro może mieć znaczenie, jeśli związek koncentruje się w bogatych w lipidy tkankach mózgowych albo jeśli aktywne metabolity współuczestniczą. To jeden z powodów, dla których nagłówki o „receptorze serotoninowym, w który trafia CBD” często wyprzedzają dane.
Drugi powód to biologia sieci. Lęk nie jest generowany przez jeden receptor. Powstaje z interakcji ciała migdałowatego, jądra łożyskowego prążka krańcowego, przyśrodkowej kory przedczołowej, hipokampa, podwzgórza i jąder pnia mózgu, w tym grzbietowego jądra szwu. CBD wydaje się przesuwać aktywność w tej sieci. Część tego przesunięcia prawdopodobnie angażuje 5-HT1A. Część może obejmować TRPV1, który przy wyższych dawkach może działać przeciwnie do anksjolizy. Część może wiązać się ze zmianami tonusu anandamidu po zahamowaniu FAAH, choć udział CBD w hamowaniu FAAH u ludzi przy dawkach terapeutycznych pozostaje dyskutowany. Część może odzwierciedlać działania przeciwzapalne lub autonomiczne, które sprzęgają się ze subiektywnym lękiem. Gdy przyjmie się taki model sieciowy, niepowodzenie jednego prostego wyjaśnienia przestaje wyglądać na słabość, a zaczyna przypominać realistyczny opis farmakologii.
To także kierunek rozwoju leków. Epoka chemii medycznej mniej interesuje się sporem, czy związek jest „jak THC”, a bardziej definiowaniem kombinacji celów i oddzielaniem pożądanych efektów od odurzenia. Logika ta pojawia się daleko poza serotoniną, od screeningu strukturalnego CB2 w artykule Journal of Medicinal Chemistry z 2016 r. „Library Docking for Cannabinoid-2 Receptor Ligands” po nowsze próby rozdzielenia analgezji od centralnego upośledzenia. Widać ją także w programach anksjolitycznych na etapie firmowym. W 2025 r. MIRA Pharmaceuticals podała, że ich kandydat MIRA-55 miał „differentiated mechanism of action” oraz „anxiolytic activity relative to THC” w danych przedklinicznych opublikowanych w komunikacie Nasdaq. Poziom dowodów musi tu pozostać wyraźny: przedkliniczny, raportowany przez firmę, nie kliniczny dowód. Mimo to sygnał jest ważny jako wskaźnik rynku i badań. Firmy aktywnie szukają związków inspirowanych cannabinoidami, które uspokajają bez działania jak THC, a mechanizmy ukierunkowane na serotoninę są częścią tych poszukiwań.
Kontekst zdrowia publicznego czyni z tego coś więcej niż spór akademicki. W 2025 r. HHS stwierdził, że 7-hydroxymitragynine „poses an imminent hazard to public safety” wspierając działania klasyfikacyjne wobec niebezpiecznych wzmacnianych produktów 7-OH. Różne modyfikacje chemiczne tworzą różne profile celu i różne ryzyka. Taka sama lekcja dotyczy przestrzeni cannabinoidów. Jeśli produkt traktuje się jako wymienny ze znanymi cannabinoidami roślinnymi tylko dlatego, że brzmi podobnie do THC lub CBD, farmakologia zostaje spłaszczona, a ocena bezpieczeństwa cierpi.
Gdzie więc lądują dowody? 5-HT1A jest najlepiej wspartym mechanizmem serotoninowym uspokajających efektów CBD, ale najsilniejsze twierdzenie, jakie obecnie wspierają dane, nie brzmi „CBD jest agonistą serotoniny”. Jest węższe i bardziej obronne: CBD często wywołuje efekty podobne do anksjolitycznych i buforujących stres, które częściowo zależą od sygnalizacji 5-HT1A, podczas gdy dokładny sposób zaangażowania wydaje się zależeć od testu, dawki, tkanki i kontekstu obwodu. To może być mniej eleganckie niż slogan o jednym receptorze. Jest też znacznie bliższe prawdy.
Poza żądaną listą: kanały sodowe i inne niekanoniczne cele już zmieniają rozmowę o bólu
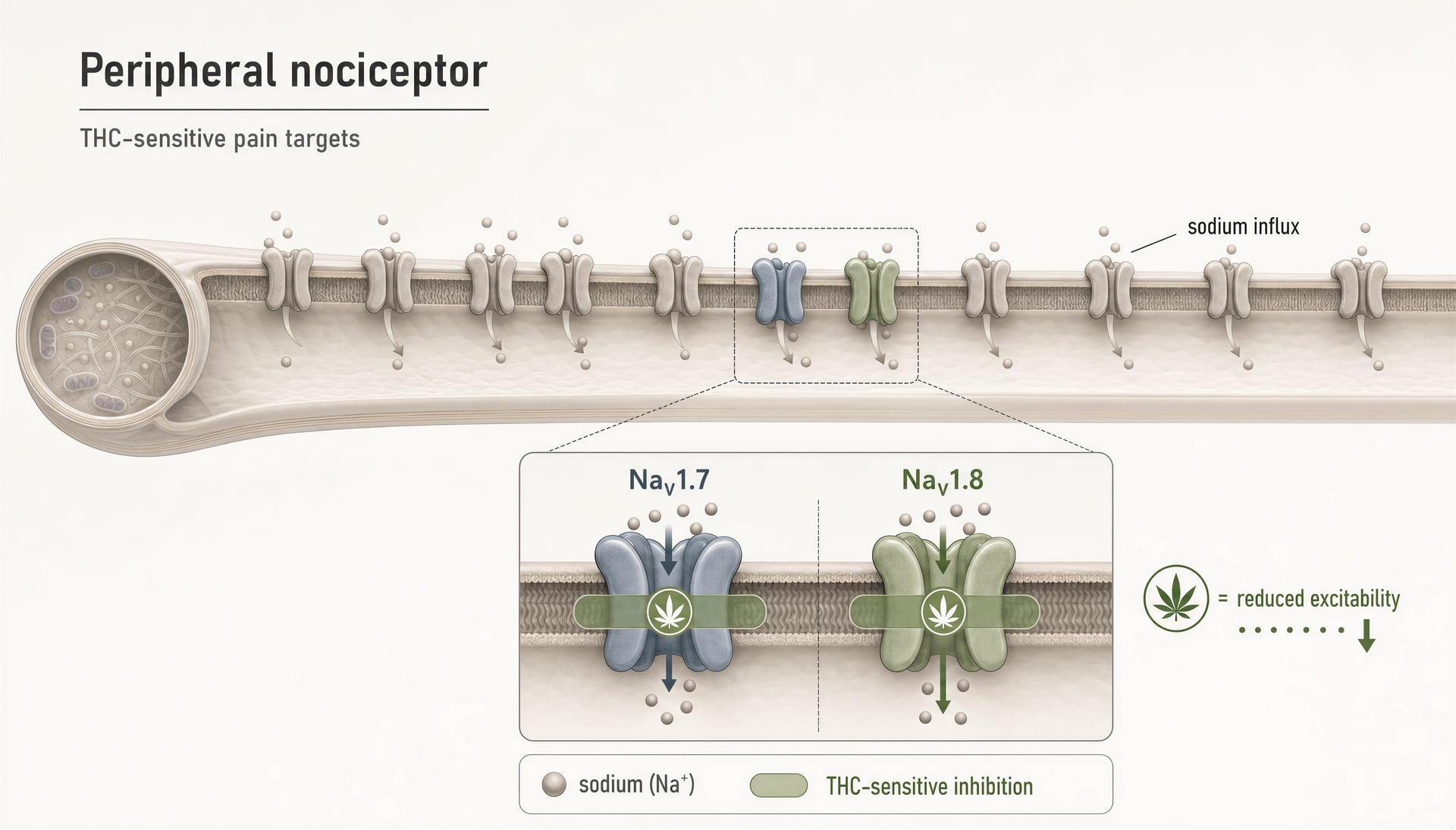
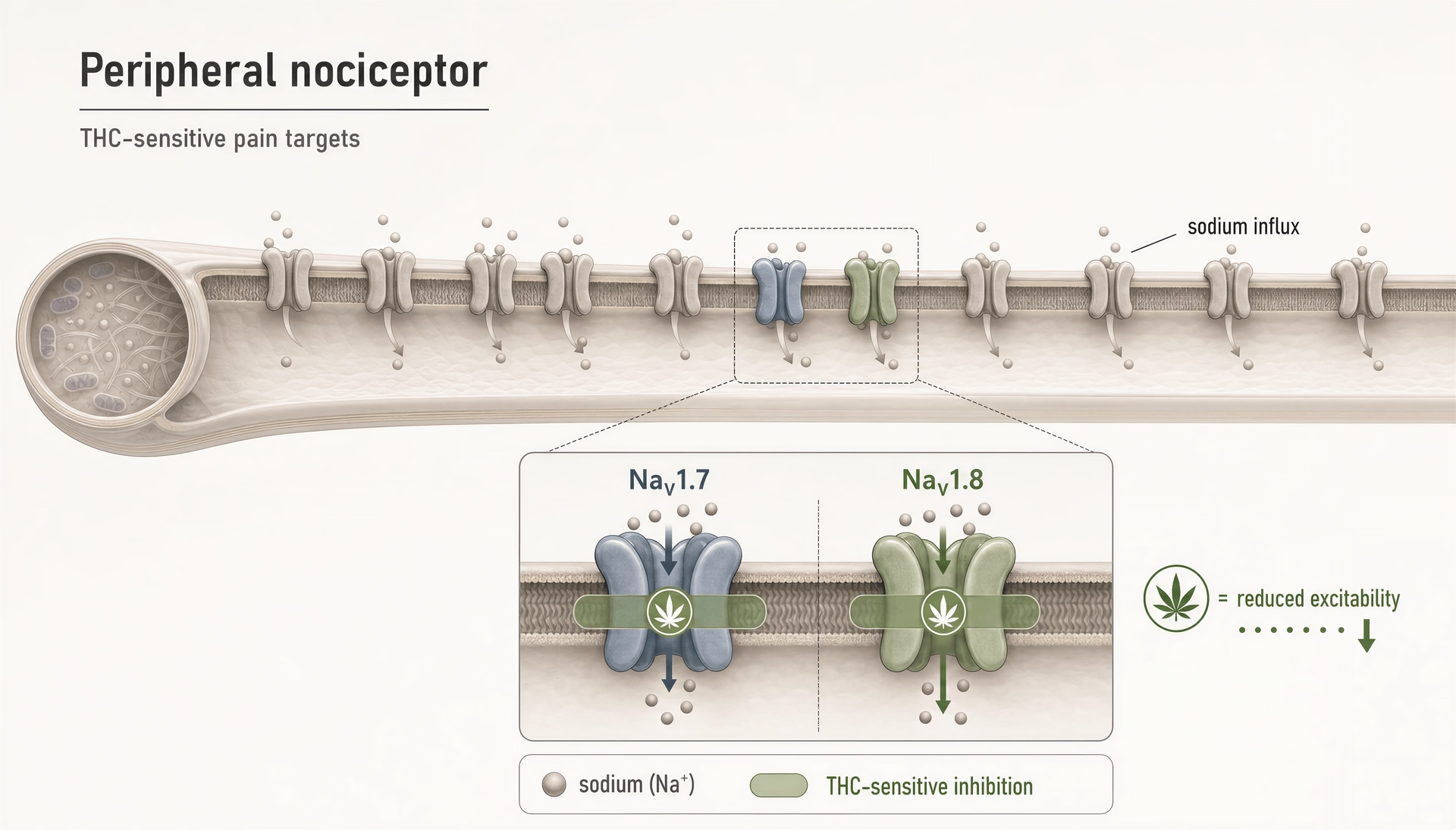
Przez lata większość publicznych dyskusji o farmakologii bólu związanej z cannabinoidami tkwiła w historii dwóch receptorów: CB1 wyjaśnia efekty psychoaktywne, CB2 efekty immunologiczne, a wszystko inne jest drugorzędne. Taki obraz jest dziś za mały. Nawet w węższym polu bólu cannabinoidy nie tylko oddziałują na kanały TRP, PPARs, GPCR-y sieroce czy szlaki związane z serotoniną. Wchodzą też w interakcje z zależnymi od napięcia kanałami sodowymi, które leżą w centrum pobudliwości nocyceptorów. To ma znaczenie, ponieważ NaV1.7 i NaV1.8 nie są pobocznymi wzmiankami; należą do najlepiej badanych molekularnych bramek sygnalizacji bólu w małych neuronach czuciowych.
Zmiana ma wymiar większy niż akademicki. Producenci leków od lat próbują blokować przewodzenie bólu na poziomie nerwów obwodowych bez odtwarzania sedacji, odurzenia, zaburzeń pamięci i potencjału nadużycia związanego z silną centralną aktywacją CB1. Jeśli cannabinoid albo szkielet wywiedziony z cannabinoidu może tłumić odpalanie nocyceptorów przez działanie na kanały NaV poza mózgiem, otwiera to zupełnie inną logikę terapeutyczną. Odsuwa rozmowę od pytania „jak silnie trafia w CB1?” ku „gdzie działa, przy jakim stężeniu i w jakiej tkance?”.
Ten szerszy maping celów pasuje także do większego momentu regulacyjnego. W 2025 r. U.S. Department of Health and Human Services ostrzegł, że „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety”, przypominając, że niewielkie zmiany chemiczne mogą dawać bardzo różną farmakologię i bezpieczeństwo. Polityka wobec cannabinoidów często nie nadąża za tym prostym faktem. Traktowanie wszystkich związków bliskich odurzeniu tak, jakby różniły się tylko pochodzeniem albo siłą równoważną THC, pomija sedno. To farmakologia na poziomie celu przewiduje efekt, ryzyko i potencjał leku.
{kind=link}
{kind=link}
{kind=link}
THC hamuje obwodowe nocyceptory poprzez oddziaływanie na kanały sodowe NaV1.7 i NaV1.8.Preliminary evidence
THC na obwodowych kanałach nocyceptywnych NaV1.7 i NaV1.8
Najbardziej bezpośrednim powodem, dla którego kanały sodowe należą dziś do każdej poważnej mapy cannabinoidów, jest raport z 2025 r. grupy Hebrew University of Jerusalem wykazujący, że THC hamuje obwodowe nocyceptory, celując w „NaV1.7 and NaV1.8 nociceptive sodium channels.” To istotne rozszerzenie słownika dziedziny. NaV1.7 i NaV1.8 są silnie eksprymowane w obwodowych neuronach czuciowych bólu, a ich rola w biologii bólu u ludzi nie jest spekulacją. Utrata funkcji NaV1.7 może powodować wrodzoną niewrażliwość na ból; mutacje zysku funkcji mogą napędzać ciężkie zespoły bólowe. NaV1.8 jest podobnie związany ze stanami bólu zapalnego i neuropatycznego, ponieważ wspiera powtarzalne wyładowania w nocyceptorach, zwłaszcza przy zdepolaryzowanych warunkach.
Zatem gdy wykazuje się, że THC hamuje te kanały, odkrycie nie trafia do szuflady „mieszane efekty poza celem”. Wskazuje na mechanizm, który może bezpośrednio zmniejszać pobudliwość włókien bólowych, zanim sygnały dotrą do rdzenia kręgowego lub mózgu.
To inna klasa mechanizmu niż lepiej znane historie o cannabinoidach. TRPV1, rozpoznany w pracach, które przyczyniły się do udziału Davida Juliusa w Nagrodzie Nobla w 2021 r., może być aktywowany lub odczulany przez kilka cannabinoidów, w tym CBD i CBG, z efektami silnie zależnymi od dawki i czasu. Sygnalizacja PPAR-gamma była przywoływana dla efektów przeciwzapalnych i metabolicznych, często z zastrzeżeniem, że znaczenie mogą mieć równie mocno kumulacja wewnątrzkomórkowa i metabolity jak cząsteczki macierzyste. GPR55 pozostaje na tyle sporny, że określanie go „CB3” nadal brzmi bardziej jak hasło niż ustalona nauka. Powiązania serotoninowe, szczególnie 5-HT1A, pomagają wyjaśnić część profilu anksjolitycznego CBD, ale obwody są zależne od kontekstu i często pośrednie. Hamowanie kanałów sodowych jest mniej efektowne. Jest też, dla bólu, potencjalnie bardziej praktyczne.
Kluczową sprawą jest tu nieswoistość farmakologiczna. Cannabinoidy są często ligandami „brudnymi” w sensie technicznym: angażują wiele celów z różnymi powinowactwami i konsekwencjami funkcjonalnymi. To nie wada nauki; to sama nauka. THC może być nadal najlepiej znany z centralnego agonizmu CB1, ale nie wyklucza to jego zdolności do modulowania obwodowych kanałów jonowych przy odpowiednich warunkach. Pytanie brzmi, czy warunki te są osiągalne in vivo w sposób pomagający pacjentom bardziej niż im szkodzący. Odkrycie Hebrew University mówi, że jest to przynajmniej na tyle prawdopodobne, by zasługiwało na poważną uwagę w rozwoju leków.
Kannabinoidowy lek przeciwbólowy może łagodzić ból bez wywoływania odurzenia dzięki mechanizmom obwodowym lub innym niż CB1.Preliminary evidence
Analgezja obwodowa bez centralnego odurzenia
Tu robi się ciekawie dla dziedziny bólu. Mechanizm cannabinoidu, który zmniejsza odpalanie obwodowych nocyceptorów, może przynajmniej teoretycznie oddzielić analgezję od zaburzeń poznawczych zwykle wiązanych z aktywacją CB1 w mózgu. To rozróżnienie stanowi centrum obecnej pracy translacyjnej, a nie poboczny bonus.
Raport ScienceDaily z 2026 r. ujął tę ideę prostym językiem: badacze zidentyfikowali „a cannabis compound that relieves pain without the high.” Tę frazę trzeba czytać ostrożnie. To sygnał na etapie badawczym, nie ustalona terapia, a popularne podsumowania często spłaszczają szczegóły mechanistyczne. Mimo to znaczenie translacyjne jest oczywiste. Jeśli aktywność przeciwbólową można generować przez ograniczenie działania obwodowego, ograniczoną penetrację mózgu, selektywne zaangażowanie celu innego niż CB1 albo kombinację tych czynników, dawny kompromis między ulgą w bólu a odurzeniem nie jest dany raz na zawsze. To problem chemii leków.
Ten punkt pomaga też wyjaśnić, dlaczego pole przesunęło się poza proste etykiety receptorowe. Artykuł ACS Journal of Medicinal Chemistry z 2016 r., „Library Docking for Cannabinoid-2 Receptor Ligands”, odzwierciedla szerszą zmianę ku projektowaniu opartemu na strukturze, zamiast traktowania cannabinoidów jako jednej rodziny farmakologicznej z jedną osią użytecznej zmienności. Chemicy pytają dziś, jak stroić kształt szkieletu, lipofilność, bias receptora, dystrybucję tkankową i los metaboliczny. Celem nie jest tylko silniejsze działanie. Celem jest selektywne działanie we właściwym miejscu.
Analgezja obwodowa jest dokładnie takim punktem końcowym, w którym te różnice mają znaczenie. Związek, który słabo przechodzi przez barierę krew–mózg, ale wyraźnie hamuje NaV1.7 lub NaV1.8 w nocyceptorach, może łagodzić ból zapalny lub neuropatyczny z dużo mniejszym odurzeniem niż sam THC. To pozostaje ambicją, nie faktem klinicznym. Jednak prace Hebrew University nadają tej ambicji uchwyt molekularny.[8]Agriculture Improvement Act of 2018. U.S. Congress. Congress.gov, 2018. https://www.congress.gov/bill/115th-congress/house-bill/2/text
To także precyzuje, jak myśleć o produktach cannabis już znajdujących się w obrocie. Definicja konopi w 2018 Farm Bill opiera się na zawartości delta-9 THC „not more than 0.3 percent on a dry weight basis.” To liczba regulacyjna, nie farmakologiczna. Nie mówi nic o kanałach sodowych, aktywacji TRP, sygnalizacji 5-HT1A, aktywnych metabolitach ani ekspozycji tkankowej. To samo dotyczy nowszych wzmacnianych lub półsyntetycznych związków odurzających. Bezpieczeństwa nie da się wywnioskować z historii pochodzenia. Trzeba je wyprowadzać z celów, stężeń i rzeczywistej farmakokinetyki.
Dlaczego te wyniki mają znaczenie dla przyszłych leków cannabinoidowych
Najsilniejsza implikacja historii NaV1.7/NaV1.8 jest taka, że przyszłe leki cannabinoidowe mogą odnieść sukces właśnie dlatego, że będą mniej „cannabinoidowe” w sensie popularnym. To znaczy, użyteczne potomki chemii cannabis mogą nie być lekami szeroko naśladującymi palone THC. Mogą to być związki, które zapożyczają część szkieletu, unikają centralnej sygnalizacji CB1 i działają zamiast tego na obwodowe kanały jonowe lub mieszane zestawy celów niekanonicznych.
Taka możliwość już pasuje do szerszej bazy dowodowej. Zatwierdzone użycie CBD dotyczy napadów, nie rutynowej analgezji, a nawet tam jego farmakologia nie jest dobrze wyjaśniona przez CB1 lub CB2. FDA label dla roztworu doustnego cannabidiol stwierdza, że jest on wskazany w napadach związanych z Lennox-Gastaut syndrome, Dravet syndrome lub complex tuberous sclerosis u pacjentów w wieku 1 roku i starszych. Innymi słowy, jedyny szeroko stosowany lek cannabinoidowy zatwierdzony przez FDA już opiera się uproszczonej narracji receptorowej. Pole bólu dopiero dogania tę samą lekcję.
Wstępne programy firmowe wskazują w tym samym kierunku, choć wymagają ostrożności. W 2025 r. MIRA Pharmaceuticals podała dane przedkliniczne, twierdząc, że MIRA-55 wykazał „differentiated mechanism of action” i „anxiolytic activity relative to THC.” Komunikaty prasowe firm nie są neutralnym dowodem, a sygnały przedkliniczne często zawodzą. Mimo to pokazują, dokąd zmierza chemia leków: od nieukierunkowanego naśladowania THC ku projektowaniu opartemu na mechanizmie.
W bólu kanały sodowe mogą stać się jedną z najbardziej znaczących gałęzi tej strategii projektowej. Nie jedyną gałęzią. TRPV1, TRPA1, PPAR-gamma, GPR55, szlaki związane z adenozyną i modulacja serotoninowa nadal pozostaną w obrazie. Ale NaV1.7 i NaV1.8 oferują coś szczególnie atrakcyjnego: bezpośredni związek z elektrycznym zachowaniem obwodowych włókien bólowych. To czyni je wyjątkowo konkretnymi celami w dziedzinie pełnej pośrednich wyjaśnień.
Efekt jest prostszy w myśleniu o cannabinoidach i bólu. Nie CB1 kontra CB2. Nie roślinne versus syntetyczne. I nie „high” versus „medical”, jakby to były kategorie molekularne. Lepszym rozróżnieniem jest mechanizm centralnego odurzenia kontra mechanizmy użyteczne obwodowo. Odkrycie Hebrew University umieszcza sam THC po obu stronach tej granicy. I właśnie dlatego ma znaczenie.
| Kannabinoid | Cele inne niż CB1/CB2 podkreślone w artykule | Główne zastrzeżenie |
|---|---|---|
| CBD | TRPV1, TRPA1, TRPM8, 5-HT1A, PPAR-gamma, GPR55, sygnalizacja związana z adenosine | Wiele sygnałów pochodzi z badań zależnych od testu i często z badań mikromolarnych |
| THC | TRPV2, zgłaszane interakcje TRPV1/TRPA1, NaV1.7, NaV1.8 | Ośrodkowe efekty CB1 mogą dominować i zaciemniać inne mechanizmy |
| CBG | TRPA1, TRPV1, TRPM8, interakcje związane z alpha-2 adrenergic i 5-HT1A | Przeniesienie stężenia in vitro na ekspozycję u ludzi jest niepewne |
| CBC | TRPA1, kanały z rodziny TRPV | Dowody u ludzi są skąpe |
| THCV | Możliwości inne niż CB1, w tym TRP i sygnalizacja metaboliczna | Dawka i kontekst mogą zmieniać pozorne zachowanie |
| CBDA / THCA | Działania związane z 5-HT1A, TRP i enzymami omawiane w pracach przedklinicznych | Stabilność, dekarboksylacja i ekspozycja komplikują interpretację |
Czym różnią się poszczególne cannabinoidy, gdy przestaje się pytać wyłącznie o CB1 i CB2
Gdy przestaje się traktować CB1 i CB2 jako całą opowieść, znany zestaw cannabinoidów wygląda znacznie mniej uporządkowanie. Te cząsteczki nie są czystymi kluczami do dwóch zamków. Są lipofilnymi związkami wrażliwymi na stężenie, które mogą trafiać w kanały jonowe, receptory jądrowe, procesy transportowe, GPCR-y sieroce, a w niektórych przypadkach w zależne od napięcia kanały sodowe. Ma to znaczenie, ponieważ ból, zapalenie, kontrola napadów, lęk i działania niepożądane często słabo mapują się na proste etykiety „agonista CB1” lub „agonista CB2”.
CBD wymusił tę zmianę bardziej niż jakikolwiek inny cannabinoid. Jego słaba klasyczna skuteczność wobec receptorów cannabinoidowych utrudniła obronę starego schematu, szczególnie wtedy, gdy FDA-approved cannabidiol oral solution uzyskał wskazania do napadów związanych z Lennox-Gastaut syndrome, Dravet syndrome i complex tuberous sclerosis u pacjentów w wieku 1 roku i starszych. Klinicznie użyteczny cannabinoid o ograniczonym odurzającym działaniu podobnym do CB1 był problemem dla redukcjonizmu receptorowego. Badacze musieli szukać dalej.
Szersze pole poszło za tym. Prace nad kanałami TRP czerpały z biologii czuciowej, która została doceniona na najwyższym poziomie, gdy Nagrodę Nobla w 2021 r. w dziedzinie fizjologii lub medycyny otrzymali David Julius i Ardem Patapoutian za odkrycie receptorów temperatury i dotyku. Farmakologia cannabinoidów przecina się z tą biologią bezpośrednio: TRPV1, TRPA1 i TRPM8 ciągle wracają, ponieważ wiele phytocannabinoids może je aktywować, hamować lub odczulać przy stężeniach istotnych eksperymentalnie. Nie znaczy to, że każdy wynik testu przewiduje efekt u człowieka. Znaczy to, że stara idea, iż „prawdziwe” działanie cannabinoidu zaczyna się i kończy na CB1/CB2, jest błędna.
CBD: prototyp złożoności poza CB1/CB2
CBD jest najlepszym przykładem tego, dlaczego nieswoistość celu ma znaczenie. Ma niskie powinowactwo i ograniczoną skuteczność wobec CB1 i CB2 w porównaniu z THC, a mimo to wyraźnie wywołuje biologicznie istotny efekt. Lukę między tymi faktami wypełniła seria hipotez roboczych, niektóre silniejsze od innych.
Kanały TRP były jedną z pierwszych poważnych alternatyw. CBD aktywuje TRPV1 w systemach heterologicznych, a TRPV1 nie jest pobocznym, egzotycznym szlakiem; to centralny kanał nocycepcji i bólu zapalnego. Aktywacja może wydawać się nieintuicyjna, jeśli celem terapii jest analgezja, ale powtarzana lub utrzymująca się aktywacja TRPV1 często prowadzi do desensytyzacji, zmniejszając późniejszą pobudliwość. To jeden z powodów, dla których farmakologia TRP może wyglądać na papierze sprzecznie. Związek może najpierw aktywować, a później wyciszać układ. CBD wykazuje też działania na TRPA1 i może hamować TRPM8 w niektórych modelach, co czyni go farmakologicznie szerszym niż zwykłe publiczne podsumowanie „nieodurzający cannabinoid”.
Historia serotoniny jest bardziej sporna, ale nadal ważna. Duża literatura przedkliniczna łączy CBD z efektami związanymi z 5-HT1A, zwłaszcza w modelach lęku, stresu i nudności. Najuczciwsze stwierdzenie nie brzmi, że CBD jest po prostu agonistą 5-HT1A o wysokim powinowactwie, jak mówi się o lekach pokrewnych buspironie, lecz że sygnalizacja 5-HT1A często uczestniczy w działaniach CBD in vivo, czasem poprzez częściowy agonizm, czasem przez mechanizmy allosteryczne lub sieciowe, które pozostają nierozstrzygnięte. To rozróżnienie ma znaczenie. Zbyt wiele podsumowań spłaszcza „uczestniczy w 5-HT1A” do „działa przez serotoninę”. Dane tego nie wspierają.
PPAR-gamma to kolejny powracający kandydat, a tu liczy się chemia wewnątrzkomórkowa. PPARs są receptorami jądrowymi, więc lipofilna cząsteczka, która rozdziela się do błon i komórek, może wpływać na nie w sposób pomijany przez model receptora powierzchniowego. CBD był raportowany jako aktywator PPAR-gamma w układach komórkowych, a sygnalizacja PPAR-gamma ma wiarygodne związki ze stanem zapalnym, metabolizmem lipidów, włóknieniem i neurozapaleniem. Jest jednak haczyk: część efektów związanych z PPAR może odzwierciedlać metabolity, dłuższą ekspozycję lub pośrednie zmiany endogennych mediatorów lipidowych, a nie jednoetapowe zajęcie receptora. Farmakologia jest wystarczająco realna, by badać ją poważnie, ale słabsza jako slogan niż jako mechanizm.
Sygnalizacja adenozynowa weszła do obrazu, ponieważ CBD wydaje się w niektórych układach hamować transport nukleozydów równowagowych, potencjalnie zwiększając pozakomórkowy ton adenozynowy i pośrednio wpływając na przeciwzapalne szlaki związane z A2A. Znowu nie jest to czysta farmakologia wiązania z receptorem. To farmakologia transportu i kontekstu tkankowego. Jeśli brzmi to bardziej chaotycznie niż „CBD trafia w CB2”, to dlatego, że tak jest. Jest też bardziej wiarygodne.
Następnie pojawia się GPR55, często wymieniany jako domniemany „CB3”. Ta etykieta nadal jest zbyt pewna siebie. CBD może antagonizować lub inaczej modulować sygnalizację związaną z GPR55 w kilku układach eksperymentalnych, a GPR55 wiązano z pobudliwością, biologią kości, zapaleniem i szlakami związanymi z nowotworami. Jednak endogenne ligandy receptora, sprzężenie szlaków i znaczenie translacyjne pozostają dyskutowane. GPR55 to użyteczny generator hipotez, a nie ustalony zamiennik CB1/CB2.
Wniosek jest prosty: CBD stał się prototypem farmakologii cannabinoidowej poza CB1/CB2, ponieważ jego klinicznie istotnych efektów nie dało się inaczej wyjaśnić. To nadal prawda.
CBG, CBC, THCV, kwaśne cannabinoidy i farmakologia minor cannabinoids
Minor cannabinoids są często sprzedawane publiczności tak, jakby każdy miał jedną cechę osobowości. Farmakologia się do tego nie stosuje.
CBG jest zwykle opisywany jako słabo aktywny wobec CB1 i CB2, ale ciekawsze sygnały leżą poza tymi receptorami. W niektórych testach oddziałuje z układami alfa-2 adrenergicznymi i 5-HT1A, wykazuje aktywność wobec kanałów TRP i był badany pod kątem efektów przeciwzapalnych i przeciwbólowych, których nie da się prosto sprowadzić do klasycznej aktywacji receptorów cannabinoidowych. Pokazuje też powracający problem: mikromolowa aktywność in vitro jest łatwa do opublikowania i trudna do przełożenia. Trafienie w receptor przy 10 mikromolach może mieć znaczenie w szalce, a nie w osoczu ludzkim — albo znaczenie wyłącznie w tkankach, gdzie związek się koncentruje.
CBC pozostawał długo słabo scharakteryzowany w porównaniu z zainteresowaniem, jakie budzi. Wydaje się, że wiarygodniej angażuje kanały rodziny TRPA1 i TRPV niż CB1, a niektóre badania sugerują przeciwzapalne lub przeciwbólowe działania u zwierząt. Istnieje też zainteresowanie jego wpływem na ton endocannabinoid system, w tym efektami mogącymi pośrednio zmieniać sygnalizację anandamidu. Ale stwierdzenie „CBC działa przez kanały TRP” jest nadal punktem wyjścia, a nie gotową odpowiedzią. Dowody u ludzi są bardzo skąpe.
THCV jest przypadkiem bardziej złożonym, ponieważ może zachowywać się inaczej zależnie od dawki i kontekstu nawet w CB1, często opisywany jako neutralny antagonista lub ligand o niskiej skuteczności przy niektórych stężeniach, a w innych jako związek bardziej agonistyczny. Poza CB1/CB2 THCV łączono z działaniem na TRP i efektami metabolicznymi, z wielokrotnym zainteresowaniem apetytem, kontrolą glikemii i bilansem energetycznym. Część tego entuzjazmu wyprzedzała dane już lata temu. Lepsza interpretacja jest taka, że THCV jest farmakologicznie interesujący właśnie dlatego, że nie mieści się w szablonie THC, a nie dlatego, że jeden wtórny cel w pełni wyjaśnił jego profil.
Kwaśne cannabinoidy zasługują na więcej uwagi niż otrzymują. THCA i CBDA często traktuje się po prostu jako „surowe” prekursory, ale ich farmakologia nie jest wyłącznie bierną gotowością do dekarboksylacji. CBDA ma jedne z ciekawszych danych dotyczących efektów przeciwwymiotnych związanych z 5-HT1A w badaniach przedklinicznych, a oba kwaśne cannabinoidy wykazywały w testach in vitro działania wobec TRP i enzymów. Ich niższa penetracja mózgu w porównaniu z neutralnymi cannabinoidami może wręcz być zaletą dla niektórych celów obwodowych lub żołądkowo-jelitowych. Problemem pozostaje jakość danych i realizm dawki. Wiele twierdzeń opiera się na skąpych badaniach na zwierzętach lub testach komórkowych o niepewnym znaczeniu dla ludzi.
Tu zderzają się polityka produktu i farmakologia. 2018 Farm Bill zdefiniował hemp jako materiał o zawartości delta-9 THC nie większej niż 0.3 percent on a dry weight basis. Ta granica prawna nie mówi nic o GPR55, TRPV1, NaV1.7, metabolitach ani analogach półsyntetycznych. Regulatorzy zaczęli reagować na tę niezgodność także w obszarach pokrewnych. W 2025 r. HHS stwierdził, że „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” wspierając działania DEA dotyczące wzmacnianych produktów 7-OH. Inna klasa leków, ta sama lekcja: zmodyfikowane lub wzbogacone produkty mogą mieć profile bezpieczeństwa ostro odbiegające od materiału źródłowego. Cannabinoidy nie są z tej logiki zwolnione.
Metabolity, claims o entourage effect i problem chemii
Jednym z powodów, dla których farmakologia cannabinoidów staje się śliska, jest to, że związek macierzysty nie zawsze odpowiada całej ekspozycji. Metabolity mogą mieć znaczenie, czasem ogromne. Publiczność nauczyła się tego głównie przez odurzające metabolity THC, ale szersza zasada dotyczy całej nauki o cannabinoidach. Droga podania, metabolizm pierwszego przejścia, dystrybucja tkankowa i różnice gatunkowe mogą zmieniać to, które cele rzeczywiście są angażowane.
Nawet THC, związek najsilniej kojarzony z CB1, nie ogranicza się do biologii CB1. W 2025 r. badacze z Hebrew University ogłosili, że THC hamuje obwodowe nocyceptory, celując w kanały sodowe NaV1.7 i NaV1.8. To ważne przypomnienie, że analgezja może zależeć od bezpośrednich efektów na aparacie pobudliwości, a nie tylko od sygnalizacji GPCR. Raport badawczy nagłośniony przez ScienceDaily w 2026 r. poszedł tą samą translacyjną drogą dalej, opisując związek pochodzący z cannabis, który łagodził ból bez odurzenia. To nadal dowód na etapie badawczym, nie ustalona medycyna, ale wskazuje na poważną strategię rozwoju leków: oddzielić analgezję od centralnego odurzenia przez wykorzystanie celów innych niż CB1, ograniczenie do obwodu albo jedno i drugie.
Chemia medyczna zmierza w tym kierunku. Screening oparty na strukturze wokół receptorów cannabinoidowych był już dobrze ugruntowany przez artykuł Journal of Medicinal Chemistry z 2016 r. „Library Docking for Cannabinoid-2 Receptor Ligands”, a nowsze wysiłki coraz częściej dążą do odrębnych mechanizmów, a nie do ogólnie „THC-like but weaker” związków. Wydany w 2025 r. komunikat MIRA Pharmaceuticals, który należy traktować jako firmowo raportowane dane przedkliniczne, a nie niezależne potwierdzenie, opisywał MIRA-55 jako związek o zróżnicowanym mechanizmie i aktywności anksjolitycznej względem THC. Ważne nie jest to, że claim jest dowiedziony. Ważne jest to, że deweloperzy leków zakładają dziś, iż separacja celów ma znaczenie.
To prowadzi nas do entourage effect. Jako wąska idea naukowa, prawdopodobne jest, że kombinacje cannabinoidów, terpenów i metabolitów mogą zmieniać farmakokinetykę albo wywoływać efekty addytywne, przeciwdziałające lub czasem superaddytywne na wielu celach. Jako ogólne wyjaśnienie, dlaczego każdy mieszany produkt cannabis rzekomo działa lepiej, jest zwykle zbyt mgliste, by je testować, i zbyt elastyczne, by je sfalsyfikować.
Problem chemiczny jest podstawowy. Mieszanka zawierająca THC, CBD, CBG, kwaśne cannabinoidy, produkty utlenione, resztkowe terpeny i zmienne metabolity nie jest jednym działaniem. To wiele ruchomych elementów, których efekty zależą od proporcji stężeń i czasu. Wiarygodna wielocechowa farmakologia istnieje. Istnieje też nieuzasadniona uproszczona narracja. To nie jest to samo.
Lepszym standardem są dowody specyficzne dla celu, powiązane z ekspozycją. Który związek, przy jakim stężeniu, w jakiej tkance, wywołuje jaki mierzalny efekt? Gdy tylko zada się te pytania, mitologia zanika, a prawdziwa opowieść o cannabinoidach staje się widoczna: nie dwa receptory, lecz zatłoczona mapa farmakologiczna.
Metody mają znaczenie: dlaczego projekt testu kształtuje to, co sądzimy o działaniu cannabinoidów
Twierdzenia o celach działania cannabinoidów często brzmią czyściej niż dane, na których się opierają. Jeden artykuł mówi, że CBD „activates TRPV1”, inny nazywa je „5-HT1A agonist”, trzeci określa GPR55 jako kandydata na receptor cannabinoidowy, a badanie dockingowe proponuje elegancką pozycję w CB2 lub w kieszeni innego GPCR-u sierotę. Wszystkie te stwierdzenia mogą być kierunkowo przydatne. Nie są jednak tym samym rodzajem dowodu.
To rozróżnienie ma znaczenie, ponieważ cannabinoidy są tłustymi, lubiącymi błony cząsteczkami, które mają złą skłonność do wyglądania na aktywne w większej liczbie miejsc, niż rzeczywiście mają znaczenie in vivo. Dziedzina nauczyła się tego boleśnie. Jeśli związek rozdziela się do błon, zmienia właściwości dwuwarstwy, gromadzi się wewnątrz komórek, tworzy aktywne metabolity albo działa dopiero przy 10–50 mikromolach, może produkować historie o celu, które rozpadają się pod ostrzejszymi warunkami. W farmakologii cannabis metody nie są technicznym dodatkiem. To one decydują, które mechanizmy przeżyją.
Testy wiązania, testy funkcjonalne i docking
Zacznijmy od najstarszego i najczystszego pytania: czy związek się wiąże? W teście wiązania radioligandowego błony lub całe komórki eksprymujące receptor inkubuje się ze znanym ligandem radioaktywnym i rosnącymi stężeniami badanego związku. Jeśli badana cząsteczka wypiera radioligand, badacze szacują powinowactwo, często raportowane jako Ki. To jest użyteczne. Ma też ograniczenia. Wiązanie mówi, że cząsteczka może zająć miejsce przy warunkach testowych; nie mówi, co dzieje się dalej.
Dlatego testy funkcjonalne mają większe znaczenie niż samo wiązanie, gdy padają twierdzenia o sygnalizacji. Dla kanałów TRP, takich jak TRPV1, TRPA1 czy TRPM8, badacze często stosują testy przepływu wapnia z barwnikami fluorescencyjnymi. Jeśli otwarcie kanału wpuszcza wapń do komórki, fluorescencja rośnie. Zaleta jest oczywista: te testy dobrze się skalują i pozwalają szybko porównać wiele związków. Problem też jest oczywisty, jeśli zna się cannabinoidy. Niektóre cannabinoidy są fluorescencyjne, niektóre zaburzają błony, niektóre pośrednio uwalniają wapń z magazynów wewnątrzkomórkowych, a niektóre wywołują desensytyzację kanału po początkowej aktywacji. Pojedynczy szczyt w krzywej wapniowej może ukrywać kilka mechanizmów.
Patch clamp jest wolniejszy, ale znacznie bardziej informatywny. Rejestruje bezpośrednio prąd jonowy. Dla kanałów jonowych, zwłaszcza kanałów sodowych takich jak NaV1.7 i NaV1.8, patch clamp może pokazać, czy lek zmienia aktywację, inaktywację, prawdopodobieństwo otwarcia lub gęstość prądu. Dlatego raport Hebrew University z 2025 r. o działaniu THC na obwodowe nocyceptory przez NaV1.7 i NaV1.8 ma znaczenie metodologiczne: bezpośrednia elektrofizjologia potrafi oddzielić rzeczywisty efekt kanałowy od mglistego sygnału komórkowego. Jeśli cannabinoid zmniejsza prąd sodowy w nocyceptorach przy stężeniach osiąganych w tkance obwodowej, ma to dla analgezji większą wagę mechanistyczną niż kolejna luźna etykieta „interakcja z receptorem”.
GPCR-y i receptory jądrowe wymagają innych odczytów. Dla GPCR-ów badacze mogą mierzyć cAMP, rekrutację beta-arrestyny, wiązanie GTPγS, fosforylację ERK albo sygnały wapniowe w komórkach inżynieryjnych. Nie są to metody wymienne. Cannabinoid może wyglądać jak agonista w jednym szlaku, częściowy agonista w innym i antagonista w trzecim. To nie jest bałagan; to bias sygnalizacyjny. Dla PPAR-gamma powszechnym podejściem jest test reporterowy, w którym promotor wrażliwy na PPAR steruje lucyferazą. Światło rośnie i związek uznaje się za aktywator PPAR. Ale testy reporterowe są o kilka kroków oddalone od bezpośredniego zaangażowania celu. Lipofilny cannabinoid może zmieniać transkrypcję pośrednio przez stres komórkowy, metabolizm lub zmianę endogennych mediatorów lipidowych.
Odczyty ekspresji genów są jeszcze dalej w dół szlaku. Jeśli praca pokazuje, że CBD zmienia transkrypty zapalne, a efekt osłabia antagonista PPAR, jest to sugestia, nie dowód rozstrzygający. Antagonista może być niedoskonały. Komórki mogą tworzyć metabolity. Cannabinoid może zmieniać ton adenozynowy, obsługę wapnia, stan redoks albo uporządkowanie błony. Mechanizm przez odejmowanie jest kruchy.
Jest jeszcze docking. Docking pyta, czy mała cząsteczka może dopasować się do modelowanej lub eksperymentalnie określonej kieszeni wiążącej i uzyskać korzystny wynik. Używany dobrze, jest triage w chemii medycznej. Używany źle, staje się dekoracyjną pewnością. Artykuł ACS Journal of Medicinal Chemistry z 2016 r., „Library Docking for Cannabinoid-2 Receptor Ligands”, jest dobrym punktem wyjścia, bo pokazuje logikę w najlepszym wydaniu: screenowanie oparte na strukturze może pomóc priorytetyzować szkielety, wskazać prawdopodobne kontakty ligand-receptor i prowadzić syntezę wokół selektywności CB2. Do tego służy docking. Generuje hipotezy. Nie dowodzi, że phytocannabinoid angażuje cel w żywej tkance, a tym bardziej nie dowodzi, że interakcja wyjaśnia analgezję, lęk czy zapalenie.
Różnice gatunkowe, metabolity i efekty błonowe
Nawet silne sygnały in vitro mogą zawieść poza szalką, ponieważ farmakologia cannabinoidów jest mocno zależna od kontekstu. Ludzkie i gryzoniowe receptory nie zawsze są funkcjonalnie identyczne. Związek aktywny wobec mysiego TRPA1 lub szczurzego odczytu związanego z 5-HT1A może mieć inną moc albo skuteczność w ludzkim ortologu. To samo dotyczy wariantów splicingowych, rezerwy receptorowej i tła komórkowego. Silnie nadekspresjonowana linia komórkowa może sprawić, że słaby ligand będzie wyglądał na istotny.
Metabolizm dodaje kolejną warstwę. Wiele cannabinoidów nie pozostaje długo w formie macierzystej. THC staje się 11-hydroxy-THC; inne struktury tworzą metabolity utlenione lub sprzężone, które mogą ostro różnić się profilem celu. Regulatorzy zwracają coraz większą uwagę na ten problem w pokrewnych sporach dotyczących polityki lekowej. W 2025 r. HHS stwierdził, że „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety”, podkreślając szerszą lekcję: wzbogacone lub uprzywilejowane metabolicznie związki odurzające mogą zachowywać się bardzo inaczej niż związek macierzysty. W przestrzeni cannabis istnieją podobne problemy z półsyntetycznymi cannabinoidami, nowymi izomerami i formulacjami zmieniającymi ekspozycję. Jeśli testuje się tylko związek macierzysty, można przeoczyć formy rzeczywiście napędzające efekt in vivo.
Błony są największym cichym czynnikiem zakłócającym. Cannabinoidy są na tyle lipofilne, że gromadzą się w dwuwarstwach i przedziałach wewnątrzkomórkowych. To oznacza, że nominalne stężenie w kąpieli jest często słabym przybliżeniem stężenia przy celu. Zastosowanie 10 mikromoli w teście komórkowym może stworzyć bardzo duże lokalne obciążenie błony, które zmienia bramkowanie kanałów lub zachowanie receptorów niespecyficznie. Może też generować fałszywie negatywne wyniki, jeśli wolne stężenie wodne jest znacznie niższe niż oczekiwano, bo związek przylega do plastiku, białek surowicy albo samej błony.
To jeden z powodów, dla których twierdzenia o wysokich stężeniach wymagają sceptycyzmu. Jeśli cannabinoid wpływa na cel dopiero powyżej 20–30 mikromolów, pierwsze pytanie powinno brzmieć: czy to rzeczywiście interakcja istotna fizjologicznie, czy raczej artefakt związany z błoną? Kanały TRP są tu szczególnie podatne na przecenienie. Są prawdziwymi celami reagującymi na cannabinoidy w pewnych przypadkach, ale efekty mogą być dwufazowe, szybko prowadzące do desensytyzacji i silnie zależne od stężenia. Krótkie wapniowe wyładowanie przy wysokich mikromolach nie oznacza automatycznie mechanizmu istotnego terapeutycznie.
Od artykułów o docking w ACS do rzeczywistej farmakologii
Chemia medyczna lubi upraszczanie, ale biologia karze za nadmierne uproszczenia. Artykuł o docking CB2 pokazuje użyteczną wersję uproszczenia: zacznij od struktury receptora, przeszukaj biblioteki, priorytetyzuj kandydatów, syntezuj analogi, testuj je w badaniach wiązania i funkcji, a potem ucz się z relacji struktura-aktywność. Taka sekwencja może budować realne leki. Może też ujawnić, że najbardziej atrakcyjna in silico pozycja należy do związku o słabej przepuszczalności, niestabilnym metabolizmie, biasie sygnalizacyjnym albo bez istotnej aktywności w komórkach natywnych.
Dystans od pozy docking do farmakologii jest dokładnie miejscem, w którym upada wiele twierdzeń o cannabinoidach. Obraz docking CBD lub CBG w TRPV1, 5-HT1A, GPR55 czy PPAR-gamma nie jest dowodem, że związek ten napędza odpowiednią fizjologię u zwierząt lub ludzi. Prawdziwa farmakologia wymaga zbieżności. Idealnie oznacza to bezpośrednie zaangażowanie celu przy wiarygodnych stężeniach, efekty funkcjonalne w układach natywnych, dane o utracie funkcji z antagonistami lub knockoutem, wsparcie farmakokinetyczne i jakiś związek z zachowaniem albo wynikiem klinicznym.
Gdy te warstwy się pokrywają, historia szybko się wzmacnia. Obecny zwrot badawczy w stronę oddzielania analgezji od odurzenia opiera się właśnie na takiej logice. Raport ScienceDaily z 2026 r. opisujący „a cannabis compound that relieves pain without the high” jest interesujący właśnie dlatego, że wykracza poza prostą równoważność THC i kieruje uwagę ku mechanizmom selektywnym wobec celu lub ograniczonym obwodowo. To samo dotyczy pracy nad kanałami sodowymi z udziałem THC. To samo dotyczy wysiłków tworzenia selektywnych ligandów poprzez docking i projektowanie strukturalne. Wzorzec jest spójny, nawet jeśli pojedyncze twierdzenia wymagają przycięcia.
To, czego prawdopodobnie nie zobaczymy w najbliższym czasie, to masowy sukces kliniczny. Nieswoistość celu, złożoność metabolitów, różnice gatunkowe i efekty formulacji ciągle psują proste historie. Ale dziedzina nie jest już uwięziona w pytaniu, czy cannabinoidy „naprawdę” chodziły o CB1 i CB2. Deweloperzy leków odpowiedzieli na to pytanie swoimi budżetami chemicznymi. Projektują przestrzeń poza tymi receptorami, bo tam dziś znajduje się najlepsza szansa na oddzielenie korzyści od obciążenia.
Poziomy dowodów: od szalki komórkowej do kliniki
Literatura o celach innych niż CB1/CB2 jest bogata, bo cannabinoidy są chemicznie nieswoiste. Jedna cząsteczka może dotknąć TRPV1, sygnalizacji związanej z 5-HT1A, PPAR-gamma, GPR55, tonus adenozynowy i zależne od napięcia kanały sodowe zależnie od stężenia, tkanki i profilu metabolitów. To daje ciekawe prace o mechanizmach. Nie czyni automatycznie dowiedzionego leku. Jeśli jest jedna zasada, która utrzymuje uczciwość tej dziedziny, brzmi ona prosto: każdy krok w górę po drabinie dowodów eliminuje dużą część twierdzeń, które na niższym szczeblu wyglądały przekonująco.
Co dowody przedkliniczne mogą, a czego nie mogą dowieść
Na dole drabiny stoją testy wiązania, rejestracje kanałów, systemy reporterowe i hodowle komórkowe. Te metody są niezbędne. Dzięki nim badacze dowiedzieli się, że farmakologia cannabinoidów wykracza daleko poza klasyczne receptory, i dlatego redukcjonistyczne wyjaśnienia cannabis wyglądają dziś na przestarzałe. Nagroda Nobla w 2021 r. uhonorowała Davida Juliusa i Ardem Patapoutiana „for their discoveries of receptors for temperature and touch”, przypominając, że biologia TRP nie jest poboczną ciekawostką; leży blisko centrum współczesnej nauki o bólu. Gdy cannabinoidy aktywują lub odczulają TRPV1, TRPA1 lub pokrewne kanały w szalce, to odkrycie ma znaczenie.
Szalka nie powie jednak, czy to samo zaangażowanie celu występuje przy tolerowanych dawkach u ludzi. Wiele efektów in vitro dla cannabinoidów pojawia się dopiero przy stężeniach mikromolarnych. Poziomy w osoczu po rzeczywistym dawkowaniu mogą być niższe, bardziej przejściowe albo przesunięte w metabolity o innej farmakologii. CBD jest tu klasycznym przykładem. Wielokrotnie pokazuje działania trudne do wyjaśnienia wyłącznie przez CB1 lub CB2, dlatego w literaturze stale pojawiają się ścieżki TRPV1, 5-HT1A, PPAR-gamma, GPR55 i adenozynowe. Jednak fakt, że CBD może modulować cel w hodowanych komórkach, nie dowodzi, że ten mechanizm napędza wynik kliniczny w epilepsji, lęku, bólu czy zapaleniu.
Modele zwierzęce są o krok bliżej medycyny, ale nadal daleko od niej. Badania na gryzoniach mogą pokazać, że cannabinoid zmniejsza allodynię, hamuje markery zapalne lub zmienia zachowanie podobne do lęku. Mogą nawet wspierać opowieści specyficzne dla celu z antagonistami, knockoutem lub ograniczeniem do obwodu. Najnowszy raport Hebrew University jest dobrym przykładem tego, dlaczego ta praca jest ekscytująca: badacze donieśli, że THC hamuje obwodowe nocyceptory, celując w kanały sodowe NaV1.7 i NaV1.8. To odkrycie przecina leniwe założenie, że analgezja związana z THC musi być historią o CB1 plus odurzenie. Jeśli efekt się potwierdzi, sugeruje działanie istotne dla bólu na poziomie samej obwodowej pobudliwości.
Nawet silna praca na zwierzętach nie dowodzi jednak, że blokada NaV1.7/NaV1.8 przez cannabinoid stanie się bezpiecznym i skutecznym ludzkim lekiem przeciwbólowym. Różnice gatunkowe mają znaczenie. Dawkowanie ma znaczenie. Droga podania ma znaczenie. Odczyty behawioralne u myszy mogą mylić. Sygnał bólowy stłumiony w preparacie nerwu lub w teście formalinowym nie musi przełożyć się na ulgę w neuropatii, chorobie zwyrodnieniowej stawów czy bólu pooperacyjnym u ludzi. Taka sama ostrożność dotyczy komunikatów firmowych. W 2025 r. MIRA Pharmaceuticals podała, że jej kandydat MIRA-55 wykazał „differentiated mechanism of action” i „anxiolytic activity relative to THC” w danych przedklinicznych. To jest dopuszczalne jako dowód na etapie badawczym. To nie jest claim terapeutyczny i nie powinno być tak traktowane.
Ta sama ostrożność dotyczy też chwytliwych nagłówków. ScienceDaily w 2026 r. nagłośniło pracę nad „a cannabis compound that relieves pain without the high.” Być może. To użyteczny kierunek projektowania leków, zwłaszcza jeśli cele obwodowe lub mechanizmy inne niż CB1 mogą oddzielić analgezję od centralnego odurzenia. Ale „without the high” w raporcie przedklinicznym jest hipotezą pod testem, a nie ustalonym faktem terapii u ludzi.
Zatwierdzony lek cannabinoidowy i wąskość obecnych wskazań
Przejdźmy na szczyt drabiny: dowody z leków zatwierdzonych. Tu pole robi się znacznie węższe. Najbardziej oczywistym przykładem w USA jest roztwór doustny cannabidiol. FDA-approved label stwierdza, że jest on wskazany „for the treatment of seizures associated with Lennox-Gastaut syndrome, Dravet syndrome, or tuberous sclerosis complex in patients 1 year of age and older.” To zdanie mówi więcej niż dziesiątki mglistych claimów wellness, ponieważ wymienia postać leku, wynik, choroby i zakres wieku.
Zauważmy, czego etykieta nie mówi. Nie mówi, że CBD jest zatwierdzony na ból, uogólniony lęk, bezsenność, nieswoiste zapalenie jelit, neuroprotekcję czy szeroką „endocannabinoid balance”. Nie potwierdza też każdego proponowanego mechanizmu z literatury przedklinicznej. Zatwierdzenie mówi nam, że określony produkt cannabidiol wykazał skuteczność i akceptowalne bezpieczeństwo w określonych zaburzeniach napadowych w kontrolowanych badaniach. Nie rozstrzyga, czy TRPV1, GPR55, 5-HT1A, PPAR-gamma, efekty wapniowe wewnątrzkomórkowe czy działanie metabolitów są dominującym klinicznym mechanizmem. Mechanizm może pozostać częściowo nierozstrzygnięty nawet wtedy, gdy skuteczność jest realna.
Ta luka między zatwierdzonym wskazaniem a folkiem mechanistycznym jest powszechna w farmakologii. Wiele leków działało u pacjentów zanim w pełni zrozumiano ich cel. Cannabinoidy nie są tu wyjątkowe. Wyjątkowe jest raczej to, jak często szerokie twierdzenia rekonstruuje się z rozproszonych wyników receptorowych, a potem mówi tak, jakby przeszły już badania kliniczne.
Wąskość obecnych wskazań ma też znaczenie dla dyskusji o zdrowiu publicznym. Regulatorzy coraz wyraźniej rozróżniają znane roślinne cannabinoidy od zmienionych, wzmacnianych lub półsyntetycznych związków odurzających o innych profilach ryzyka. W 2025 r. HHS stwierdził, że „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” wspierając działania klasyfikacyjne wobec wzmacnianych produktów 7-OH. Oświadczenie to dotyczyło produktów związanych z kratomem, a nie cannabis, ale lekcja polityczna pozostaje ta sama: gdy producenci zaczynają wzbogacać, modyfikować lub syntetyzować silnie psychoaktywne cząsteczki, skróty myślowe oparte na pochodzeniu roślinnym stają się zawodne. Farmakologia na poziomie celu zaczyna mieć duże znaczenie.
Dlaczego historie o mechanizmie często wyprzedzają dane kliniczne
Wyprzedzają, bo historie mechanistyczne są szybkie, wyraziste i publikowalne. Kliniczny dowód jest wolny i kosztowny. Praca komórkowa może pokazać, że cannabinoid aktywuje TRPA1, antagonizuje GPR55 albo zmienia sygnalizację 5-HT1A w projekcie trwającym miesiące. Przekonujące randomizowane badanie bólu przewlekłego może trwać latami i mimo to zakończyć się niepowodzeniem, bo wielkość efektu jest mała, działania niepożądane ograniczają dawkę albo cel przedkliniczny nigdy nie był realnie angażowany u pacjentów.
Chemia cannabinoidów sprzyja też nadinterpretacji. Strukturalnie podobne związki mogą mieć bardzo różne profile celu, a metabolizm po podaniu może jeszcze ten obraz przebudować. Droga podania zmienia historię ponownie. Dawkowanie doustne daje metabolizm pierwszego przejścia; inhalacja zmienia kinetykę; formulacje miejscowe lub obwodowe mogą faworyzować cele lokalne zamiast centralnych. Nawet kategoria prawna „hemp” mówi farmakologicznie niewiele. 2018 Farm Bill wyznaczył granicę na poziomie delta-9 THC „not more than 0.3 percent on a dry weight basis”, czyli na poziomie ustawowym, nie biologicznym.
Prawidłowe odczytanie dowodów poza CB1/CB2 nie polega więc ani na odrzuceniu, ani na hype. Literatura przedkliniczna naprawdę pokazuje, że cannabinoidy działają na więcej niż CB1 i CB2. Zwłaszcza w bólu kanały TRP i kanały sodowe zasługują na poważną uwagę. W przypadku CBD cele nieklasyczne nie są pobocznymi przypisami; prawdopodobnie stanowią rdzeń wyjaśnienia jego profilu. Ale prawdopodobieństwo nie jest skutecznością, zaangażowanie celu nie jest korzyścią dla pacjenta, a zatwierdzenie jednego leku cannabinoidowego do niewielkiej grupy zaburzeń napadowych nie ratyfikuje znacznie większej chmury twierdzeń zbudowanej z diagramów receptorowych, zachowania myszy i wyników z szalki.
Bezpieczeństwo, regulacje i dlaczego farmakologia działań poza celem ma znaczenie dla zdrowia publicznego
Problemy zdrowia publicznego nie układają się w prosty sposób według diagramów receptorowych. Cannabinoid może pochodzić z rośliny, z konopi, być półsyntetyczny albo całkowicie syntetyczny, a mimo to generować ryzyka słabo przewidywane przez etykietę, pochodzenie czy kategorię prawną. To praktyczne znaczenie farmakologii poza celem. Gdy cząsteczka dotyka kanałów TRP, receptorów serotoninowych, PPARs, systemów sygnalizacji podobnych do GPR55, kanałów sodowych i innych celów spoza CB1/CB2, profil bezpieczeństwa może się zmieniać w sposób istotny dla nadzoru nad zatruciami, standardów produktu, polityki dotyczącej prowadzenia pojazdów pod wpływem i ryzyka uzależnienia.
Błąd nie jest tylko naukowy. Jest regulacyjny. Polityka wobec cannabis często traktowała „cannabinoid” tak, jakby był już wyjaśnieniem.
Wzmacniane intoxicants i lekcja polityczna płynąca z 7-OH
Najwyraźniejsze obecne ostrzeżenie nie pochodzi nawet z klasycznego składnika cannabis. W 2025 r. U.S. Department of Health and Human Services stwierdził, że „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” wspierając działania DEA dotyczące wzmacnianych produktów 7-OH. To zdanie ma znaczenie, ponieważ pokazuje, że federalne organy zdrowia wyznaczają granicę między znaną ekspozycją botaniczną a skoncentrowanymi lub chemicznie manipulowanymi związkami odurzającymi, które mogą zachowywać się bardzo inaczej w rzeczywistych populacjach.
Lekcja polityczna przenosi się bezpośrednio na cannabinoidy. Szeroka kategoria, taka jak „hemp-derived”, „plant-based” czy nawet „cannabinoid-like”, mówi regulatorom niemal nic o tym, jakie cele angażuje związek przy stężeniach istotnych dla użycia. Nie mówi też konsumentom prawie nic o sile działania, początku, czasie trwania, ryzyku interakcji czy potencjale nadużycia.
Produkty wzbogacone w 7-OH stały się problemem zdrowia publicznego, ponieważ chemia zmieniła ekspozycję. Gdy rynek przechodzi od śladowej naturalnej obecności do skoncentrowanego składnika aktywnego, farmakologia przestaje być ciekawostką i staje się główną kwestią bezpieczeństwa. Taki sam wzorzec pojawił się już wokół półsyntetycznych i strukturalnie zmienionych cannabinoidów sprzedawanych pod parasolem legalności hemp po 2018 Farm Bill zdefiniował hemp przez próg delta-9 THC „not more than 0.3 percent on a dry weight basis.” Ten próg suchej masy definiuje uprawę, nie farmakologię. Nie eliminuje aktywności wobec TRP, blokady kanałów sodowych, sygnalizacji 5-HT1A, efektów GPR55 ani metabolitów o dłuższej trwałości i innej penetracji OUN.
Dlatego działania poza celem nie są pobocznym przypisem. To jedna z dróg, przez które produkty marketingowo opisane jako bliskie cannabis mogą generować nieoczekiwane szkody. Związek przedstawiany publicznie jako „THC-like but legal” może różnić się od THC w intrinsic efficacy wobec CB1, ale może też różnić się w mniej widoczny sposób: silniejszą stymulacją układu sercowo-naczyniowego, większym potencjałem prodrgawkowym lub anksjogennym, cięższą dysforią, nietypową sedacją albo toksycznością kształtowaną przez metabolizm, a nie sam związek macierzysty. Te możliwości nie są hipotetyczne w abstrakcyjnym sensie; właśnie dlatego agencje zdrowotne reagują inaczej na wzmacniane intoxicants niż na szerokie kategorie roślinne.
Sensowna odpowiedź polityczna zaczyna się od pytań na poziomie celu. Jakie receptory i kanały są angażowane? Przy jakich stężeniach? W jakich tkankach? Jakie są główne metabolity? Czy istnieją dowody na hamowanie kanałów sodowych, które może zmieniać nocycepcję lub przewodzenie w sercu? Czy aktywacja TRPV1 prawdopodobnie odczuli ból, czy raczej podrażni i pogorszy objawy przy niższych lub krótkich ekspozycjach? To trudniejsze pytania niż „czy to cannabinoid”, ale są właściwe.
Granice myślenia o THC-equivalence
THC-equivalence jest atrakcyjna, bo upraszcza prawo, opodatkowanie, etykietowanie i rozmowy o upośledzeniu. Jest też często błędna. Dwa związki mogą wywoływać pewien stopień odurzenia, a mimo to ostro różnić się ryzykiem lęku, działaniem psychotomimetycznym, efektem przeciwbólowym, odpowiedzią tętna, kontrolą wymiotów, rozwojem tolerancji i obciążeniem odstawiennym, ponieważ nie dzielą tej samej szerszej mapy celów.
Nawet sam THC nie jest farmakologicznie wyczerpany przez CB1 i CB2. W 2025 r. badacze z Hebrew University donieśli, że THC hamuje obwodowe nocyceptory, celując w NaV1.7 i NaV1.8. To uderza w proste opowieści „THC działa tu, CBD tam”. Jeśli kanoniczny psychoaktywny cannabinoid może bezpośrednio wpływać na zależne od napięcia kanały sodowe związane z bólem, bezpieczeństwa i skuteczności nie da się wyprowadzać wyłącznie z marki cannabinoidu. Dawka, droga podania, dystrybucja i ekspozycja tkankowa stają się decydujące.
Ta sama myśl pojawia się z odwrotnej strony przy CBD. Jego zatwierdzony roztwór doustny jest wskazany przez FDA w napadach związanych z Lennox-Gastaut syndrome, Dravet syndrome lub complex tuberous sclerosis u pacjentów w wieku 1 roku i starszych, co samo w sobie nie było dobrze wyjaśnione przez agonizm CB1, ponieważ CBD nie jest prostym odurzającym agonistą CB1. Jego profil od dawna kieruje badaczy ku innym mechanizmom, w tym TRPV1, 5-HT1A, sygnalizacji związanej z adenozyną i PPAR-gamma. Nie wszystkie te mechanizmy są równie dobrze ugruntowane u ludzi, ale razem pokazują jedną rzecz nie do pominięcia: efekty cannabinoidów często siedzą w sieci, a nie w jednym przełączniku receptora.
Perspektywa sieciowa wyjaśnia też, dlaczego odurzenie jest złym nadrzędnym miernikiem bezpieczeństwa publicznego. Produkt może być mniej odurzający niż THC, a mimo to powodować niepokojące interakcje lekowe, wpływ na enzymy wątrobowe, obciążenie układu sercowo-naczyniowego, reakcje paniczne lub sedację. Może też być bardziej odurzający, ale nie bardziej przewidywalny. Publiczność często słyszy „słabszy niż THC” albo „silniejszy niż THC”, jakby to rozstrzygało sprawę. Nie rozstrzyga. Zwykle są to stwierdzenia o jednym wyrazistym fenotypie, a nie o pełnym profilu toksykologicznym.
Pipeline badawczy już wychodzi poza THC-equivalence. Komunikat Nasdaq MIRA Pharmaceuticals z 2025 r. opisywał dane przedkliniczne dla MIRA-55, twierdząc, że wykazuje on „differentiated mechanism of action” i aktywność anksjolityczną względem THC. Komunikaty firmowe są słabszym dowodem niż recenzowane dane kliniczne, więc claim należy traktować ostrożnie. Mimo to kierunek jest realny: chemia medyczna próbuje oddzielić pożądane efekty od centralnego odurzenia przez zmianę zaangażowania celu. Tę samą logikę translacyjną widać w raporcie ScienceDaily z 2026 r. opisującym pracę nad „a cannabis compound that relieves pain without the high.” Wyniki na etapie badawczym nie są dowodem klinicznym, ale wzmacniają centralny punkt regulacyjny. Jeśli korzystne efekty można oddzielić od odurzenia, to szkody można odurzeniu także oddzielić. Produkt low-high nie jest automatycznie produktem low-risk.
Dlaczego nowe cannabinoidy wymagają skrupulatnej oceny na poziomie celu
Nowe cannabinoidy wymagają więcej, a nie mniej, skrupulatności właśnie dlatego, że często trafiają na rynek zanim ich farmakologia zostanie zmapowana u ludzi. Dawny skrót polegał na pytaniu, czy cząsteczka wiąże CB1 lub CB2. Lepsze pytanie brzmi, co jeszcze robi i czy te działania stają się istotne przy realnych dawkach po inhalacji, podaniu doustnym lub metabolizmie.
W bólu oczywistymi przykładami są kanały TRP i kanały sodowe. David Julius i Ardem Patapoutian otrzymali Nagrodę Nobla w 2021 r. w dziedzinie fizjologii lub medycyny za odkrycia receptorów temperatury i dotyku, przypominając, że biologia czuciowa opiera się na celach, na które cannabinoidy mogą wpływać poza klasycznymi receptorami cannabinoidowymi. Aktywacja TRPV1 może prowadzić do analgezji przez desensytyzację, ale może też powodować podrażnienie i jest silnie zależna od stężenia. Regulator patrzący tylko na aktywność CB1 przeoczy cały ten ośrodek ryzyka i korzyści.
W bezpieczeństwie psychiatrycznym znaczenie ma sygnalizacja serotoninowa. CBD było wielokrotnie omawiane jako kandydat anksjolityczny związany z 5-HT1A, ale udział serotoniny może być pośredni i zależny od kontekstu. Ta niepewność nie jest powodem do ignorowania celu; jest powodem, by badać go starannie przed normalizacją produktów jako nieszkodliwych. To samo dotyczy GPR55, czasem przedstawianego jako kandydat „CB3” mimo trwającego sporu. Sporny cel nadal jest celem, który może kształtować sygnalizację wapniową, pobudliwość i odpowiedzi zapalne.
W efektach metabolicznych i zapalnych cele wewnątrzkomórkowe, takie jak PPAR-gamma, jeszcze bardziej komplikują obraz. Sygnalizacja przez receptory jądrowe nie wygląda jak szybki efekt odurzający, ale może być ważna dla przewlekłej ekspozycji, dystrybucji w tkance tłuszczowej, zmian transkrypcyjnych i interakcji z innymi stanami chorobowymi. Komunikacja publiczna budowana wokół pytania, czy coś „gets you high”, pomija te wolniejsze, cichsze ryzyka.
Tu regulacja, toksykologia i komunikacja bezpieczeństwa konsumenckiego potrzebują literaturoznawstwa molekularnego. Nie każdy hit in vitro będzie miał znaczenie kliniczne. Różnice gatunkowe są realne. Metabolity mogą dominować. Agencje powinny jednak wymagać paneli celów, charakterystyki metabolitów, danych dawka-odpowiedź i badań u ludzi istotnych dla upośledzenia, zanim założą, że jeden nowy cannabinoid można regulować tak samo jak inny. Artykuł ACS Journal of Medicinal Chemistry z 2016 r. „Library Docking for Cannabinoid-2 Receptor Ligands” pokazuje, jak dziś działa odkrywanie leków: projektowanie oparte na strukturze, inżynieria selektywności i wyraźna uwaga na różnice na poziomie receptora. Regulacja powinna przestać udawać, że rynek jest prostszy niż wie już chemia medyczna.
Zdrowie publiczne nie może sobie pozwolić na redukcjonizm receptorowy. „Cannabinoid” to etykieta wyjściowa. Nie jest wnioskiem o bezpieczeństwie.
Odkrywanie leków: projektowanie cannabinoidów i cząsteczek inspirowanych cannabinoidami dla celów innych niż CB1/CB2
Odkrywanie leków wokół cannabinoidów dawno wyszło poza stare pytanie, czy cząsteczka jest „CB1-active” lub „CB2-active”. Uproszczenie to zawsze było kruche, ponieważ wiele związków związanych z cannabinoidami jest farmakologicznie nieuporządkowanych: trafiają w kanały jonowe, GPCR-y spoza klasycznej pary, wewnątrzkomórkowe receptory jądrowe, procesy transportowe i enzymy metabolizujące, często z różnymi efektami zależnymi od stężenia, tkanki i drogi podania. Dla chemików medycznych ten chaos nie jest tylko problemem. To także szansa.
Główny cel projektowy jest dostatecznie jasny: zachować działanie przeciwbólowe, przeciwzapalne lub anksjolityczne, a jednocześnie zmniejszyć obciążenie związane z silną aktywacją CB1 w mózgu. Obciążenie to nie jest abstrakcją. Sedacja, upośledzenie poznawcze, odurzenie, potencjał nadużycia i ograniczające dawkę efekty psychiatryczne są właśnie powodem, dla którego „THC-like” jest często złym profilem w programie rozwojowym, nawet jeśli sam THC wykazuje użyteczną farmakologię. Obecny klimat regulacyjny wzmacnia ten punkt. W 2025 r. HHS poparł działania klasyfikacyjne, stwierdzając, że „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety”, przypominając, że zmodyfikowanych lub wzbogaconych intoxicants nie można zakładać, że zachowują się tak samo jak znane związki roślinne tylko dlatego, że są blisko na półce marketingowej. Farmakologia na poziomie celu ma znaczenie.
Ograniczenie do obwodu, selektywność funkcjonalna i bias sygnalizacyjny
Jednym ze sposobów uniknięcia działań niepożądanych w OUN jest metoda prosta, ale skuteczna: utrzymać lek poza mózgiem. Ograniczenie do obwodu można projektować przez zwiększenie polarności powierzchni, zwiększenie zdolności do wiązań wodorowych, zmianę pKa albo uczynienie cząsteczki substratem transporterów efflux w barierze krew–mózg. Sama idea nie jest wyjątkowa dla nauki o cannabinoidach, ale w tej dziedzinie pasuje szczególnie dobrze, ponieważ sygnalizacja bólu często zaczyna się w obwodowych nocyceptorach, tkankach objętych zapaleniem i zwojach korzeni tylnych.
To właśnie tam cele spoza CB1/CB2 stają się szczególnie atrakcyjne. Raport Hebrew University z 2025 r. argumentował, że THC hamuje obwodowe nocyceptory poprzez NaV1.7 i NaV1.8, dwa główne czynniki pobudliwości nocyceptywnej. Jeśli ten mechanizm się potwierdzi w różnych układach, ma to ogromne znaczenie. NaV1.7 od dawna uchodzi za cel premium w bólu, ponieważ ludzkie mutacje utraty funkcji w SCN9A mogą powodować głęboką wrodzoną niewrażliwość na ból. Szkielet cannabinoidowy, który zachowuje modulację kanałów sodowych na obwodzie, a jednocześnie minimalizuje centralną sygnalizację CB1, nie byłby po prostu „mniej odurzającym THC”. Byłby innym rodzajem leku przeciwbólowego.
Kanały TRP tworzą podobną szansę. Szersza biologa czuciowa została doceniona najwyższym wyróżnieniem, gdy Nagrodę Nobla w 2021 r. w dziedzinie fizjologii lub medycyny otrzymali David Julius i Ardem Patapoutian za odkrycie receptorów temperatury i dotyku. TRPV1, TRPA1 i pokrewne kanały są głęboko związane z nocycepcją i sygnalizacją zapalną, a kilka phytocannabinoids oddziałuje z nimi w sposób zależny od stężenia i czasem paradoksalny: początkowa aktywacja może być następnie zastąpiona desensytyzacją, co może przyczyniać się do analgezji. To utrudnia chemię medyczną, a nie ułatwia. Ale też oznacza, że związek nie musi być czystym ortosterycznym ligandem CB receptorowym typu on-off, by mieć wartość terapeutyczną.
Selektywność funkcjonalna dodaje jeszcze jedną warstwę. Nawet w samych CB1 lub CB2 ligandy mogą preferować jeden wynik sygnałowy nad innym, przesuwając równowagę między szlakami białka G, rekrutacją beta-arrestyny, internalizacją receptora i downstream programami transkrypcyjnymi. Mówiąc prościej, dwie cząsteczki mogą obie „wiązać CB1”, a mimo to zachowywać się w żywej tkance bardzo różnie. Artykuł z 2016 r. w Journal of Medicinal Chemistry, „Library Docking for Cannabinoid-2 Receptor Ligands”, odzwierciedla, jak dalece strategia projektowania przesunęła się ku strukturze zamiast ku prostym etykietom receptorowym. Ta sama logika obejmuje dziś także receptory spoza CB: chemicy chcą szkieletów, których kształt, lipofilność i ograniczenia konformacyjne kierują je ku mechanizmowi istotnemu dla bólu lub lęku, a z dala od ciężkiego balastu centralnego sygnalizowania.
CBD jest stałym dowodem na to, że lek związany z cannabinoidami może mieć znaczenie kliniczne bez wyjaśniania przez agonizm CB1/CB2. Etykietowanie FDA dla roztworu doustnego cannabidiol, zaktualizowane w 2024 r., obejmuje napady związane z Lennox-Gastaut syndrome, Dravet syndrome i complex tuberous sclerosis u pacjentów w wieku 1 roku i starszych. Niezależnie od tego, jaki zestaw efektów TRPV1, 5-HT1A, GPR55, adenozynergicznych, wewnątrzkomórkowych i sieciowych ostatecznie wyjaśnia tę skuteczność, nie jest to prosta historia CB1. Projektanci leków to zauważyli.
MIRA-55 i dążenie do odrębnych mechanizmów
MIRA-55 jest użytecznym studium przypadku nie dlatego, że cokolwiek rozstrzyga, ale dlatego, że pokazuje, jak firmy dziś opisują programy cannabinoidowe. W komunikacie przekazanym przez Nasdaq w 2025 r. MIRA Pharmaceuticals podała, że ich kandydat wykazał „differentiated mechanism of action” i „anxiolytic activity relative to THC” w pracy przedklinicznej. To sformułowanie mówi niemal wszystko o obecnym sygnalizowaniu wobec inwestorów i regulatorów. Bycie inspirowanym cannabinoidami nie wystarcza. Firma chce dystansu od prostego naśladowania THC, zwłaszcza w takich wskazaniach jak lęk, gdzie centralne działania niepożądane mogą zniweczyć korzyść.
Mimo to ostrożność jest obowiązkowa. „Differentiated mechanism” w komunikacie firmowym to claim, nie wniosek. Może oznaczać zmienione zaangażowanie receptora, inną dystrybucję tkankową, odmienne metabolity, częściowy agonizm, bias funkcjonalny, działania na kanały poza celem albo po prostu inny profil behawioralny w jednym teście zwierzęcym. Bez pełnych paneli farmakologicznych, krzywych dawka-odpowiedź, identyfikacji metabolitów, danych o zajęciu receptorów i replikacji w warunkach zaślepionych, to raczej hipoteza niż wynik.
Mimo to strategia stojąca za claimem jest wiarygodna. Jeśli związek potrafi zmniejszyć zachowanie podobne do lęku przy mniejszym odurzeniu, zaburzeniu pamięci lub hamowaniu lokomocji niż THC, chemia medyczna prawdopodobnie zmieniła jedną lub więcej z czterech rzeczy: penetrację mózgu, intrinsic efficacy wobec CB1, zaangażowanie celów innych niż CB, takich jak 5-HT1A lub kanały TRP, albo konwersję metaboliczną do aktywnych form o innym profilu celu. To właśnie osie, na których dziś konkurują programy cannabinoidowe.
MIRA-55 pokazuje też problem dekonwolucji celu. Cząsteczki podobne do cannabinoidów są często farmakologicznie „brudne”. To nie jest ocena moralna; to ostrzeżenie mechanistyczne. Jeśli pojawia się sygnał anksjolityczny przedkliniczny, nie można zakładać, że wyjaśnia go jeden receptor. Sygnalizacja serotoninowa może być bezpośrednia lub pośrednia. Efekty PPAR-gamma mogą wymagać akumulacji wewnątrzkomórkowej lub metabolitów. GPR55 może wyglądać na ważny w jednym teście, a marginalny w innym. Efekt in vitro przy 10 mikromolach może być nieistotny, jeśli wolne stężenia w mózgu nigdy nie zbliżają się do tego poziomu in vivo.
Jak wygląda realistyczny pipeline cannabinoidowy
Realistyczny pipeline jest węższy i bardziej zdyscyplinowany niż sugeruje publiczna retoryka. To nie parada „non-psychoactive cannabis compounds” pędzących ku zatwierdzeniu. To filtr.
Na wejściu stoją modyfikacja szkieletu i triage: klasyczne cannabinoidy, nieklasyczne cannabinoidy, lipidy inspirowane endocannabinoidami i niezależne chemotypy naśladujące jedną użyteczną cechę farmakologii cannabinoidowej bez przejmowania całego pakietu. Chemicy zmieniają długość łańcucha bocznego, ograniczenia pierścieniowe, stereochemię, pozycję heteroatomów i miejsca słabe metabolicznie, a następnie testują nie tylko CB1 i CB2, ale też TRPV1, TRPA1, kanały NaV, wybrane GPCR-y sierocę i testy związane z serotoniną. Następnie kandydaci przechodzą badania ADME, analizę stężenia niezwiązanego i pomiary brain-to-plasma. Wiele z nich odpada właśnie tam.
Programy, które przechodzą dalej, zwykle dzielą się na kilka wiarygodnych kategorii. Jedna to analgetyki ograniczone obwodowo, gdzie marzeniem jest uśmierzenie bólu przez kanały sodowe, desensytyzację TRP, modulację zapalenia albo obwodową sygnalizację cannabinoidową bez znacznej ekspozycji OUN. Druga to ligandy CB1 o biasie lub niskiej skuteczności, które próbują zachować sygnalizację terapeutyczną przy mniejszym odurzeniu. Trzecia to związki wielomechanizmowe, często bardziej realistyczne niż czysto jednocelowe podejście, gdzie umiarkowane działania w kilku węzłach bólowych lub lękowych przewyższają „czysty”, ale klinicznie słaby ligand.
Translacyjny hak, który utrzymuje tę dziedzinę przy życiu, jest prosty: oddzielenie analgezji od odurzenia wydaje się możliwe, przynajmniej w systemach badawczych. Podsumowanie ScienceDaily z 2026 r. opisujące nowe badanie „a cannabis compound that relieves pain without the high” należy czytać ostrożnie, bo nagłówki wyprzedzają dane, ale koncepcja pasuje do ogólnego kierunku chemii medycznej. Tak samo jak praca o kanałach sodowych dla THC. Tak samo jak wysiłki budowania selektywnych ligandów przez docking i projektowanie sterowane strukturą. Wzorzec jest spójny, nawet gdy poszczególne twierdzenia wymagają korekty.
To, czego pipeline prawdopodobnie nie daje w krótkim terminie, to masowego sukcesu klinicznego. Nieswoistość celu, złożoność metabolitów, różnice gatunkowe i wpływ formulacji ciągle psują proste historie. Ale dziedzina nie stoi już w miejscu, pytając, czy cannabinoidy naprawdę chodziły o CB1 i CB2. Deweloperzy leków odpowiedzieli już na to pytanie swoimi budżetami chemicznymi. Projektują przestrzeń poza tymi receptorami, bo tam dziś leży najlepsza szansa na oddzielenie korzyści od obciążenia.
Powszechne nieporozumienia i nierozstrzygnięte spory
Największym błędem w publicznej dyskusji o cannabinoidach jest redukcjonizm receptorowy: potrzeba wtłoczenia chaotycznej farmakologii w jeden nagłówkowy cel. Ten nawyk zawsze był kruchy, a staje się coraz bardziej ryzykowny, gdy regulatorzy i deweloperzy leków mierzą się ze związkami, które nie są już po prostu „THC” lub „CBD” w prostym, pochodzącym z roślin sensie. W 2025 r. HHS powiedział, że „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” wspierając działania DEA wobec wzmacnianych produktów 7-OH. Inna klasa leków, ta sama lekcja: gdy chemicy modyfikują, wzbogacają lub półsyntetyzują szkielet odurzający, stare założenia o działaniu receptorowym i bezpieczeństwie mogą szybko zawieść. Nauka o cannabis ma ten sam problem. Cannabinoid nie jest magicznym kluczem do jednego zamka. Zwykle jest ligandem o szerokim spektrum, którego efekty w realnym świecie zależą od stężenia, ekspozycji tkankowej, metabolizmu i tego, które cele poza głównymi stają się przy tych poziomach istotne.
Czy naprawdę istnieje receptor CB3
Nie ma konsensualnego receptora CB3.
To brzmi ostro, ale dowody tego wymagają. Przez lata proponowano kilka receptorów jako kandydatów na „CB3”, zwłaszcza GPR55, czasem GPR18 i okazjonalnie inne GPCR-y sierocę, które w niektórych układach testowych reagują na ligandy cannabinoidowe lub lipidy związane z endocannabinoid system. Ale proponowany cel nie jest akceptowaną klasą receptorową. CB1 i CB2 zdobyły swoje nazwy dzięki zbieżnym dowodom: klonowaniu, powtarzalnej farmakologii ligandów, dystrybucji tkankowej, sygnalizacji i szerokiej replikacji. Kandydaci na CB3 nigdy nie przeszli tej poprzeczki.
GPR55 jest zwykle głównym podejrzanym. Jest eksprymowany w mózgu, komórkach odpornościowych, jelicie i kości, a niektóre cannabinoidy rzeczywiście z nim oddziałują. CBD często opisywano jako antagonistę GPR55 w testach komórkowych; aktywność wykazują też niektóre syntetyczne cannabinoidy. Jednak farmakologia jest niespójna między laboratoriami, ligandami i odczytami. Niektóre związki wyglądają aktywnie w jednym teście sygnalizacyjnym i cicho w innym. Różnice gatunkowe komplikują obraz. Endogenne ligandy są dyskutowane. Najważniejsze jednak, że nazwanie GPR55 „CB3” sugeruje ustalone miejsce w kanonicznej rodzinie receptorów cannabinoidowych, którego dziedzina mu nie przyznała.
Ma to znaczenie, ponieważ etykiety mogą wyprzedzać dowody. Gdy receptor dostanie chwytliwy pseudonim, pseudonim zaczyna wykonywać pracę wyjaśniającą, na którą nie zapracował. Ból? CB3. Lęk? CB3. Efekty kostne? CB3. To nie jest farmakologia; to branding. Ostrożniejsze stanowisko mówi, że GPR55, GPR18, GPR119 i pokrewne receptory są celami spoza CB1/CB2 o różnym stopniu dowodów na modulację przez cannabinoidy lub lipidy podobne do cannabinoidów. Niektóre mogą mieć ogromne znaczenie w konkretnych tkankach. Żaden nie uzyskał konsensusu jako „trzeci receptor cannabinoidowy”.
Chemia medyczna dostosowała się do tego faktu. Artykuły chemiczne nie zachowują się tak, jakby jedna nowa nazwa receptora miała naprawić system. Artykuł Journal of Medicinal Chemistry z 2016 r. „Library Docking for Cannabinoid-2 Receptor Ligands” dobrze pokazuje, gdzie naprawdę jest dziedzina: projektowanie oparte na strukturze, selektywność receptorowa, inżynieria szkieletu i optymalizacja ukierunkowana na cel, a nie mitologia o tajemniczym CB3, który wyjaśni wszystko. Podobnie wypowiedzi firm o kolejnych generacjach związków inspirowanych cannabinoidami coraz częściej podkreślają mechanizmy różnicujące, a nie po prostu silniejszy agonizm receptorowy. Komunikat prasowy MIRA Pharmaceuticals z 2025 r. dotyczący MIRA-55 wyraźnie twierdził, że kandydat ma „differentiated mechanism of action” oraz aktywność anksjolityczną względem THC. To materiał promocyjny, nie ustalona nauka, ale odzwierciedla realną zmianę strategiczną: użyteczne terapie cannabinoidowe mogą powstać przez odejście od brutalnego odurzenia CB1, a nie przez odkrycie jednego receptora uniwersalnego.
Czy CBD działa głównie przez serotoninę
Też nie, choć sygnalizacja serotoninowa jest częścią historii.
W kulturze popularnej CBD bywa przedstawiane tak, jakby było zasadniczo naturalnym lekiem 5-HT1A. To uproszczenie przetrwało, bo ma za sobą realne dowody. W badaniach przedklinicznych CBD wykazywał efekty podobne do anksjolitycznych i przeciwstresowych, które w niektórych paradygmatach były zmniejszane przez antagonistów 5-HT1A. Badania eksperymentalne u ludzi także sugerowały, że mechanizmy serotoninowe mogą uczestniczyć w ostrych efektach anksjolitycznych w pewnych warunkach. Ale „uczestniczą” nie znaczy „działa głównie przez”.
CBD jest farmakologicznie szerokie. Ma niskie powinowactwo do CB1 i CB2 w porównaniu z THC, ale nie czyni go to automatycznie pojedynczym lekiem serotoninowym. W literaturze możliwe składniki efektu obejmują TRPV1, 5-HT1A, GPR55, sygnalizację adenozynową, PPAR-gamma, gospodarkę wapniową wewnątrzkomórkową, ton endocannabinoid system związany z FAAH w niektórych kontekstach i działanie metabolitów, które mogą nie odpowiadać cząsteczce macierzystej. Stężenie ma tu większe znaczenie, niż wielu autorów popularnych wyjaśnień przyznaje. Efekt receptora widziany in vitro przy stężeniach mikromolarnych może nie dominować u ludzi po standardowej dawce doustnej, gdzie wchłanianie jest zmienne, a metabolizm pierwszego przejścia znaczny.
Rejestr kliniczny działa przeciw twierdzeniom jednomechanistycznym. Najsilniej rozpoznane przez FDA zastosowanie oczyszczonego CBD nie dotyczy lęku, lecz napadów: roztwór doustny jest wskazany w Lennox-Gastaut syndrome, Dravet syndrome i complex tuberous sclerosis u pacjentów w wieku 1 roku i starszych. Już samo to mówi coś ważnego. Gdyby CBD „działało głównie przez serotoninę”, jego najbardziej powtarzalny profil terapeutyczny trudno byłoby pogodzić z tym, do czego używają go klinicyści. Serotonina może mieć znaczenie w stanach lękowych, szczególnie w odpowiedziach związanych z 5-HT1A, ale farmakologia CBD u ludzi nie redukuje się do etykiety serotoninowej.
Nawet w lęku mechanizm prawdopodobnie zmienia się wraz z dawką i kontekstem. TRPV1 jest dobrym przykładem. CBD może aktywować TRPV1, a sygnalizacja TRP jest na tyle centralna dla biologii czuciowej, że David Julius i Ardem Patapoutian otrzymali Nagrodę Nobla w 2021 r. za odkrycia receptorów temperatury i dotyku. Jednak efekty TRPV1 nie są liniowe. Aktywacja może być następnie odczulona; niskie i wysokie dawki mogą dawać różne rezultaty behawioralne. Więc gdy ktoś mówi „CBD działa przez serotoninę”, właściwa odpowiedź brzmi: częściowo, czasem i prawdopodobnie nie samodzielnie.
Czy jeden cel może wyjaśnić efekt całej rośliny
Nie, żaden pojedynczy cel nie może wyjaśnić efektu całej rośliny cannabis, a próba wymuszenia takiego wyjaśnienia zwykle zaciemnia więcej, niż wyjaśnia.
Efekty całej rośliny wynikają z nałożonych zmiennych. Zacznij od składu: THC, CBD, minor cannabinoids, takich jak CBG, CBC, THCV, kwaśne prekursory, produkty utlenienia i metabolity mają różne profile celu. Dodaj drogę podania, a obraz znów się zmienia. Wdychane cannabinoidy szybko docierają do mózgu; produkty doustne przechodzą metabolizm pierwszego przejścia i tworzą inne aktywne formy. Prawo USA nadal definiuje hemp przez próg delta-9 THC „not more than 0.3 percent on a dry weight basis”, ale ta granica prawna niewiele mówi o farmakologii wszystkiego innego w próbce ani o tym, co powstaje po metabolizmie.
Dodaj potem swoistość tkankową. Cannabinoid może równocześnie wpływać na centralny CB1, obwodowe kanały TRP, sygnalizację immunologiczną i receptory jądrowe. Raport Hebrew University z 2025 r., że THC hamuje obwodowe nocyceptory przez NaV1.7 i NaV1.8, jest tu ostrym przykładem, bo rozbija leniwe równanie „efekt THC=efekt CB1.” Jeśli nawet THC ma znaczące działania na kanały sodowe związane z bólem, to idea, że kwiat, ekstrakt lub produkt spożywczy ma jeden nadrzędny receptor, staje się trudna do obrony. Badania z 2026 r. nagłośnione przez ScienceDaily — cannabis compound łagodzący ból bez odurzenia — zmierzają w tym samym kierunku, choć pozostają na etapie badawczym. Analgezja może być oddzielona od centralnego odurzenia przez wykorzystanie ograniczenia do obwodu lub celów spoza CB1. To właśnie kierunek, w którym zmierza dziedzina.
Znaczenie mają także oczekiwania i biologia indywidualna. Wcześniejsze doświadczenia, poziom lęku, genetyka, płeć, aktywność enzymów wątrobowych, sen, stan zapalny i współstosowane leki wszystko przesuwają to, jak dany produkt jest odczuwany i co robi fizjologicznie. Terpeny mogą w niektórych przypadkach wnosić udział, ale nie należy ich traktować jak magicznych dyrygentów całego efektu. Ich stężenia są często niskie, a dowody u ludzi słabsze niż sugeruje język marketingowy.
Trudniejszy, ale dokładniejszy pogląd brzmi tak: efekty cannabis są emergentne. Powstają z wielu umiarkowanych interakcji, nie z jednego sloganowego receptora. To czyni naukę mniej uporządkowaną. Czyni ją też uczciwszą.
Praktyczna interpretacja dla czytelników, klinicystów i badaczy
Praktyczna lekcja płynąca z farmakologii cannabinoidów poza CB1/CB2 jest prosta, ale wymagająca: twierdzenia mechanistyczne powinny być poddawane ostrzejszej kontroli, a nie łatwiejszej akceptacji, gdy artykuł odwołuje się do kanałów TRP, PPARs, GPR55, 5-HT1A, sygnalizacji adenozynowej lub kanałów NaV. Cannabinoidy są często farmakologicznie nieswoiste. To może być użyteczne w odkrywaniu leków. Może też mylić czytelników, którzy traktują każdy hit receptora w szalce jako wyjaśnienie efektu klinicznego.
Dobrą zasadą jest ranking dowodów według odległości od pacjenta. Test wiązania to początek, nie koniec. Następne są badania sygnalizacji komórkowej. Praca na zwierzętach może doprecyzować prawdopodobieństwo. Eksperymentalna farmakologia u ludzi jest ważniejsza. Dowody z leków zatwierdzonych są najważniejsze, a nawet tam etykieta może nie rozstrzygać mechanizmu. Roztwór doustny cannabidiol, na przykład, jest zatwierdzony przez FDA w napadach związanych z Lennox-Gastaut syndrome, Dravet syndrome i complex tuberous sclerosis u pacjentów w wieku 1 roku i starszych, ale jego profil terapeutyczny nie jest jasno wyjaśniony przez aktywację CB1 lub CB2. Ta luka właśnie dlatego sprawia, że twierdzenia o TRPV1, GPR55, 5-HT1A i celach wewnątrzkomórkowych wciąż powracają.
Jak krytycznie czytać twierdzenia o mechanizmie działania cannabinoidów
Zacznij od gatunku i układu. Czy efekt wykazano w tkance ludzkiej, linii komórkowej gryzonia, oocytach Xenopus czy nadekspresjonowanych receptorach w HEK293? To nie są układy wymienne. GPR55 jest tu dobrym przykładem: jedno badanie może wykazać, że CBD zachowuje się jak antagonista w układzie rekombinowanym, podczas gdy inny kontekst daje słabe lub zmienne sygnały, bo różnią się ekspresja receptora, endogenne lipidy i projekt testu. Nazywanie tego „mechanizmem” jest zwykle przedwczesne.
Potem pytaj o stężenie. Tu wiele twierdzeń o cannabinoidach się rozpada. Artykuł może raportować aktywację TRPV1, transaktywację PPAR-gamma lub hamowanie kanałów sodowych przy stężeniach mikromolarnych. Dobrze. Ale czy omawiana dawka u człowieka rzeczywiście tworzy wolne stężenia tkankowe w tym zakresie? Doustne cannabinoidy mają pierwszy metabolizm przejściowy, duże wiązanie z białkami i nierówną dystrybucję tkankową. Trafienie w cel przy 30 mikromolach in vitro może być ciekawą chemią i nieistotną farmakologią przy łóżku pacjenta. Z drugiej strony może się zdarzyć odwrotnie: lokalna akumulacja tkankowa, aktywne metabolity lub rozdział do lipidów mogą sprawić, że cel wewnątrzkomórkowy będzie bardziej prawdopodobny, niż sugerują same poziomy osoczowe. Tak czy inaczej, stężenie nie jest kwestią uboczną. Jest kwestią główną.
Liczy się także bezpośredni pomiar celu. Czy badacze rzeczywiście zablokowali efekt selektywnym antagonistą, wyciszyli receptor, czy zmierzyli prąd kanałowy? A może wyprowadzili mechanizm z podobieństwa do wcześniejszej literatury? Dla kanałów TRP, zwłaszcza TRPV1 i TRPA1, ma to znaczenie, ponieważ aktywacja może być dwufazowa i prowadzić do desensytyzacji. Związek może najpierw aktywować kanał, a potem zmniejszyć downstream responsiveness, co oznacza, że „agonista” nie zawsze mapuje się prosto na „więcej bólu” albo „więcej odczucia ciepła”. To jeden z powodów, dla których Nagroda Nobla 2021 dla Davida Juliusa i Ardem Patapoutiana, przyznana „for their discoveries of receptors for temperature and touch,” jest tu tak istotna: sygnalizacja czuciowa jest mechanistycznie bogata, a uproszczone etykiety receptorowe często zawodzą.
Historia NaV pokazuje, jak wygląda mocniejszy dowód. Badacze Hebrew University poinformowali w 2025 r., że THC hamuje obwodowe nocyceptory, celując w NaV1.7 i NaV1.8. To twierdzenie jest bardziej informatywne niż mgliste stwierdzenia, że THC „działa poza CB1”, ponieważ nazywa kanały związane z bólem, już centralne dla badań nad analgezją. To także przedefiniowuje stare założenie. Nawet związek znany z centralnego odurzenia CB1 może mieć klinicznie istotne działania nie-CB, szczególnie w tkance obwodowej.
Czytelnik powinien też uważać na konkurencyjne wyjaśnienia. Załóżmy, że badanie łączy CBD z anksjolizą przez 5-HT1A. Rozsądna hipoteza. Ale czy wykluczono sedację? Czy modulacja CB1 była pośrednia? Czy eksperyment rozróżniał efekt receptorowy od zmian tonusu endocannabinoid system, wychwytu adenozyny, sygnalizacji zapalnej albo oczekiwań u ludzi? Leki wielocechowe rzadko same ogłaszają, który cel wykonuje większość pracy w danym modelu.
Obecny klimat regulacyjny czyni taki sceptycyzm czymś więcej niż akademickim. W 2025 r. HHS stwierdził, że „7-hydroxymitragynine (7-OH) poses an imminent hazard to public safety” wspierając działania klasyfikacyjne wobec wzmacnianych produktów 7-OH. To nie przypadek cannabis, ale lekcja jest jasna: regulatorzy coraz częściej rozróżniają znane składniki roślinne od wzbogaconych, półsyntetycznych lub inaczej zmodyfikowanych intoxicants o innych profilach mocy i bezpieczeństwa. Polityka wobec cannabinoidów, która traktuje wszystkie związki jak skalowane wersje delta-9-THC, jest farmakologicznie przestarzała.
Jakie pytania klinicyści powinni zadawać o znaczenie celu
Klinicyści nie muszą znać wszystkich sierocych GPCR-ów, by dobrze interpretować twierdzenia o cannabinoidach. Potrzebują zdyscyplinowanej listy kontrolnej.
Po pierwsze: czy efekt wykazano u ludzi, czy tylko w komórkach i na zwierzętach? Model zapalny u myszy może wspierać prawdopodobieństwo udziału PPAR-gamma lub TRPA1, ale nie ustala korzyści u pacjenta. Po drugie: przy jakich stężeniach lub dawkach? Jeśli proponowany cel jest angażowany tylko przy poziomach wyższych niż osiągane przez standardowe dawkowanie doustne, mechanizm może nie wyjaśniać rutynowych wyników klinicznych. Po trzecie: czy cel mierzono bezpośrednio? Zajęcie receptora, elektrofizjologia, odwrócenie przez antagonistę lub zaburzenie genetyczne są warte więcej niż narracyjna inferencja.
Po czwarte: czy droga podania czyni claim bardziej czy mniej wiarygodnym? Inhalacja, podanie doustne, transdermalne i miejscowe tworzą bardzo różne profile ekspozycji. Mechanizm przeciwbólowy ograniczony do obwodu jest bardziej wiarygodny dla związku miejscowego lub obwodowo ograniczonego niż dla cząsteczki, która szybko zalewa mózg. To rozróżnienie ma znaczenie, jeśli celem terapeutycznym jest analgezja bez odurzenia.
Po piąte: czy istnieją konkurencyjne wyjaśnienia związane ze znanymi działaniami niepożądanymi? Jeśli pacjent zgłasza mniejszy lęk po preparacie cannabinoidowym, czy jest to efekt anksjolityczny zależny od 5-HT1A, zmniejszenie bólu, nieswoista sedacja, czy oczekiwanie? Jeśli markery zapalne się zmieniają, czy głównym czynnikiem jest PPAR-gamma, czy może zaszły szersze zmiany metaboliczne lub immunologiczne wcześniej? Mechanizmu nie powinno się wnioskować wyłącznie z poprawy objawów.
Dla klinicystów minor cannabinoids wymagają tej samej ostrożności. CBC, CBG, THCV i kwaśne cannabinoidy bywają omawiane tak, jakby każdy miał stabilny podpis celu. Literatura nie uzasadnia jeszcze takiej pewności. Niektóre są obiecujące. Żaden nie powinien być traktowany jako farmakologicznie rozstrzygnięty tylko dlatego, że marka lub wątek w mediach społecznościowych mówi o swoistości receptora.
Dokąd najpewniej zmierza dziedzina
Najbardziej wiarygodnym kierunkiem krótkoterminowym jest analgezja obwodowa. Atrakcyjność translacyjna jest oczywista: oddzielić ulgę w bólu od centralnego odurzenia. Raport ScienceDaily z 2026 r. opisujący pracę nad „a cannabis compound that relieves pain without the high” należy czytać jako sygnał badawczy, nie gotową odpowiedź kliniczną, ale dobrze oddaje kierunek, w którym celuje chemia medyczna. To samo dotyczy pracy Hebrew University nad NaV1.7/NaV1.8 z 2025 r.: biologia bólu pcha naukę o cannabinoidach ku nerwom obwodowym, kanałom jonowym i ekspozycji ograniczonej do tkanki.
Ligandy receptorów jądrowych o działaniu przeciwzapalnym to kolejna mocna ścieżka. Twierdzenia wokół PPAR-gamma potrzebują lepszych danych o zaangażowaniu celu u ludzi, ale koncepcja jest na tyle wiarygodna, by uzasadniać poważny rozwój, szczególnie tam, gdzie przecinają się szlaki metaboliczne i zapalne. Wielecechowe anksjolityki również prawdopodobnie pozostaną aktywnym obszarem. MIRA Pharmaceuticals podała w komunikacie Nasdaq z 2025 r., że ich kandydat MIRA-55 wykazał „differentiated mechanism of action” i aktywność anksjolityczną względem THC w danych przedklinicznych. Ponieważ to dane raportowane przez firmę i przedkliniczne, należy je traktować ostrożnie. Mimo to odzwierciedlają realny trend: badacze nie są już zadowoleni z pytania, czy kandydat jest „jak THC” lub „jak CBD”. Chcą określonych profili celu.
Ta sama zmiana jest widoczna w chemii medycznej. Artykuł Journal of Medicinal Chemistry z 2016 r. „Library Docking for Cannabinoid-2 Receptor Ligands” sygnalizował podejście oparte na strukturze, które od tego czasu tylko się rozwinęło: budować ligandy dla konkretnych celów, konkretnych tkanek i konkretnych biasów sygnalizacyjnych. Lepiej zdefiniowane minor cannabinoids prawdopodobnie wyłonią się z takiego myślenia, a nie z szerokich twierdzeń, że każdy rzadki cannabinoid ma unikalną niszę wellness.
Praktyczny wniosek jest prosty. Czytając farmakologię cannabinoidów, za każdym razem zadawaj pięć pytań: Czy efekt wykazano u ludzi czy tylko w komórkach? Przy jakich stężeniach? Czy cel mierzono bezpośrednio? Czy dawka rzeczywiście dociera do tego celu w żywej tkance? Czy istnieją konkurencyjne wyjaśnienia? Przyszłość nauki o cannabinoidach to nie mniej specyficzna farmakologia, lecz więcej.
Źródła
- [1]HHS and FDA support DEA action on dangerous 7-OH products. HHS Press Room, 2025. https://www.hhs.gov/press-room/hhs-fda-support-dea-7-oh-scheduling.html
- [2]EPIDIOLEX (cannabidiol) oral solution label. FDA drug label, 2024. https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/210365s016lbl.pdf
- [3]Library Docking for Cannabinoid-2 Receptor Ligands. Journal of Medicinal Chemistry, 2016. https://pubs.acs.org/doi/10.1021/acs.jmedchem.6c00835
- [4]MIRA Pharmaceuticals Reports New Preclinical Data Demonstrating MIRA-55's Differentiated Mechanism of Action and Anxiolytic Activity Relative to THC. Nasdaq press release, 2025. https://www.nasdaq.com/press-release/mira-pharmaceuticals-reports-new-preclinical-data-demonstrating-mira-55s
- [5]Psychoactive cannabinoid THC inhibits peripheral nociceptors by targeting NaV1.7 and NaV1.8 nociceptive sodium channels. Research portal summary, 2025. https://cannabinoids.huji.ac.il/publications/psychoactive-cannabinoid-thc-inhibits-peripheral-nociceptors-targeting
- [6]A cannabis compound that relieves pain without the high. ScienceDaily, 2026. https://www.sciencedaily.com/releases/2026/06/260619033343.htm
- [7]The Nobel Prize in Physiology or Medicine 2021. Nobel Prize Press Release, 2021. https://www.nobelprize.org/prizes/medicine/2021/press-release/
- [8]Agriculture Improvement Act of 2018. Congress.gov, 2018. https://www.congress.gov/bill/115th-congress/house-bill/2/text